Welcome to the Meggers Laboratory!
Asymmetric Catalysis - Synthetic Methods - Sustainable Chemistry - Catalyst Design
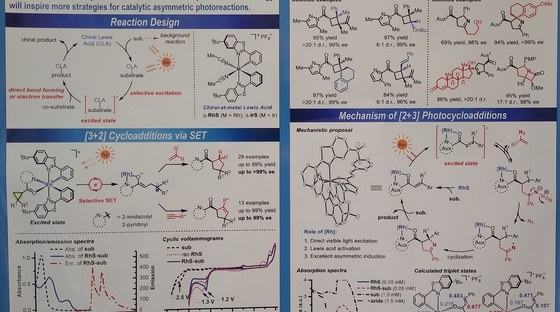
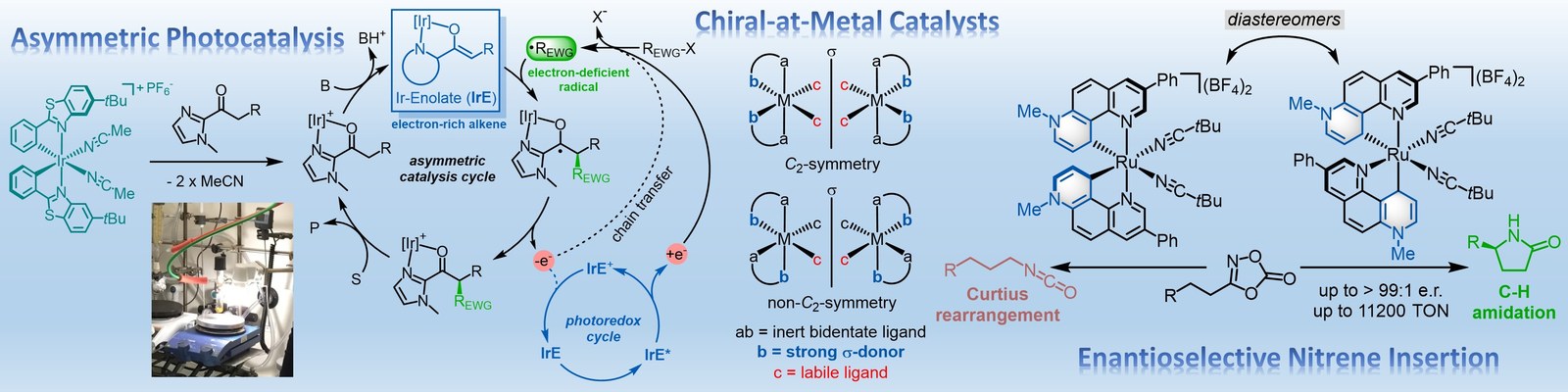
The Meggers laboratory is developing novel strategies for asymmetric catalysis. For example, we have pioneered the general use of "chiral-at-metal" catalysts in which the metal center both serves as the exclusive stereogenic center and the same time acts as the reactive center for catalysis. Such catalysts have been applied to asymmetric photocatalysis, asymmetric electrochemistry and enantioselective nitrene chemistry. Supported by an ERC Advanced Grant, we recently also started a research program on sustainable asymmetric iron catalysis.