Institut für Zytobiologie
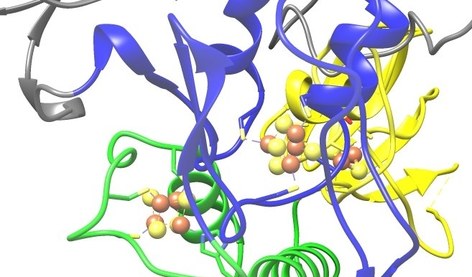
Unser Institut ist Teil des Fachbereichs Medizin und des Zentrums für Synthetische Mikrobiologie (SYNMIKRO) der Philipps-Universität Marburg. Es übernimmt Aufgaben in der zellbiologischen / biochemischen Grundlagenforschung und in der Ausbildung von Studierenden der Humanmedizin, Zahnmedizin und Humanbiologie. Zentrale Forschungsvorhaben widmen sich den molekularen Mechanismen der Biogenese und Funktion von Eisen-Schwefelproteinen und dem intrazellulären Proteintransport.