Hauptinhalt
Neuronal cell death / Neurodegenerative disorders
Research area and techniques
Mechanisms of neuronal cell death and brain damage
- Pathogenetic mechanisms in neurodegenerative diseases (stroke, Alzheimer's disease, epileptic seizures, brain trauma)
- Neuronal apoptosis: initial molecular signaling pathways and gene expression
- Mitochondrial mechanisms of neuronal cell death
- Generation of reactive oxygen species (ROS) and oxidative stress-related signaling
- DNA damage-induced signalling pathways
- Caspase-independent Apoptosis; Apoptosis inducing factor (AIF)
- Cellular mechanisms of brain damage: disturbance of the neurovascular unit and related inflammatory processes
Mechanisms of neuroprotection
- Inhibition of pro-apoptotic bcl-2 family proteins
- Protection of mitochondrial function
- Inhibition of AIF-mediated cell death signalling
- Neuroprotection by p53-inhibitors
- Activation of endogenous pathways of neuroprotection by activation of NF-kappa B
- Inhibition of oxidative stress and related signalling pathways
- Vector systems for efficient siRNA delivery to the CNS
Experimental techniques
- Primary cultures of hippocampal and cortical neurons (mice, rats), cell lines (HT-22 cells, PC12 cells)
- Models of neuronal cell death relevant for neurodegenerative diseases
- Immunhistochemistry, immuncytochemistry, ELISA, immunoblots, immunprecipitation, confocal laser scanning microscopy
- RT-PCR, TUNEL, antisense oligonucleotides; RNAi-technology
- Evaluation of mitochondrial function and levels of reactive oxygen species by using fluorescent dyes
- Confocal microscopy of cultured neurons, time lapse in vivo imaging
- Electromobilityshift assays (NF-kB, p53)
- Enzyme activity assays
- Animal models of cerebral ischemia (mice, rats)
- Stereotactic intracerebral injections/infusions
- siRNA vectors
Several prominent neurological disorders including stroke and Alzheimer's disease (AD) manifest symptoms that result from degeneration and death of neurons. Although the genetic and environmental factors that initiate neurodegeneration differ among diseases, a shared biochemical cascade of events appears to carry out the cell death process. This cascade involves increased oxidative stress, disruption of calcium homeostasis and activation of a death program called apoptosis. Neuronal apoptosis involves mitochondrial ion permeability changes, cytochrome c release and activation of caspases and caspase-independent mechanisms such as the mitochondrial release of apoptosis inducing factor (AIF). An important regulatory step in apoptosis occurs at mitochondrial membranes where members of the Bcl-2 family of proteins either promote (Bax, Bid) or prevent (Bcl-2, Bcl-xl) membrane permeability transition.
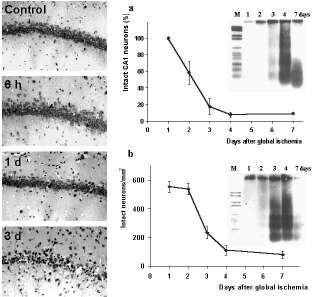
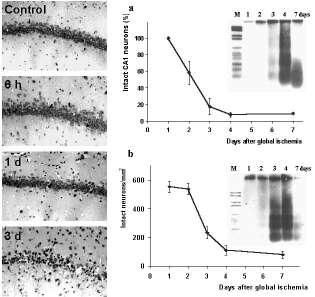
Figure 1. Delayed neuronal cell death after global cerebral ischemia.
{kind=link}
Photomicrographs of Nissl-stained brain sections show the delay of neuronal damage in CA1 subfield of the hippocampus after 10 min transient forebrain ischemia in the rat Panels on the right show the quantification of surviving neurons in the vulnerable regions of the hippocampus (a) and the dorsolateral striatum (b) after ischemia. Note that the delayed increase of apoptotic DNA-laddering after transient forebrain ischemia as detected by electrophoresis is consistent with the timecourse of neuronal cell loss in both brain regions. larger view
Our research over the last years in primary neuronal cultures, cell lines and rodent models of cerebral ischemia focussed on early signaling events in apoptosis in order to define promising targets for new therapeutic strategies of neuroprotection. Signaling events that act upstream of mitochondrial changes in neuronal apoptosis are not fully established but may include DNA damage initiating the activation of the tumor suppressor protein p53.
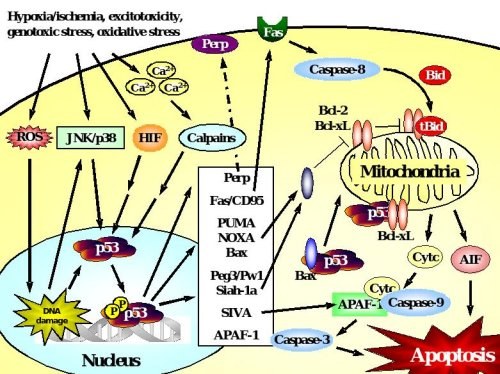
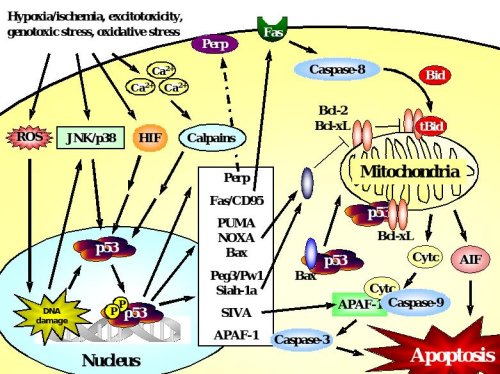
Figure 2. p53-dependent cell death in neurons.
{kind=link}
Accumulation and transcriptional activation of p53 occurs rapidly in response to a wide variety of insults including DNA damage, oxidative stress, metabolic compromise or excitotoxicity. Depending on the initial stress stimulus activation of p53 occurs via various pathways that may intersect with each other upstream of p53 activation. p53 exerts its deadly function by transactivation of pro-apoptotic target genes including those encoding Bax, the BH3-only proteins PUMA and Noxa, which translocate to mitochondria where they mediate disruption of the mitochondrial membrane potential and release of apoptotic factors including cytochrome c and AIF. Many transcriptional targets such as Peg3/Pw1, Siah-1a and SIVA act in a similar way by interacting with pro-apoptotic members of the Bcl-2 family at the level of mitochondria. In addition, p53 may promote cell death via transactivation of the death receptor Fas or upregulation of APAF-1 which promotes caspase-dependent apoptosis after formation of the apoptosome with cytochrome c and caspase-9. In addition to such transcriptional control of the cell death machinery, p53 can directly trigger apoptosis after translocation to mitochondria, a process that can occur in synapses (synaptic apoptosis) and may involve interactions with Bax or Bcl-xL.
Although DNA damage is widely considered as a late event in final stages of apoptosis, our data now imply a role for DNA damage and related signaling, such as activation of p53 or ataxia telangiectasia mutated (ATM) in early stages of neuronal apoptosis. Recently, we described a neuroprotective role for the DNA repair enzyme DNA-dependent protein kinase (DNA-PK) in neurons. Neurons lacking DNA-PK were highly susceptible to various insults in vitro and in vivo, exposing DNA repair as an essential mechanism of endogenous survival signaling. Using the p53 inhibitor pifithrin-alpha (PFT) and other newly synthesized analogues we could demonstrate the essential role of p53 in various in vitro and in vivo models relevant to neurodegenerative disorders including stroke, brain trauma, epilepsy and Alzheimer's disease. The latest results exposed reversible inhibition of p53 as a promising therapeutic strategy in neurological disorders, because p53 inhibitors block the apoptotic cascade and concomitantly enhance endogenous protective signaling through NF-kB, even if administered up to 6 h after ischemia or brain trauma.
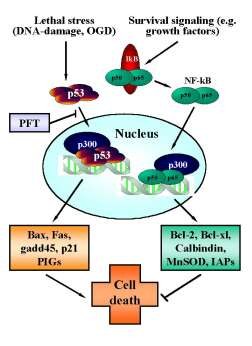
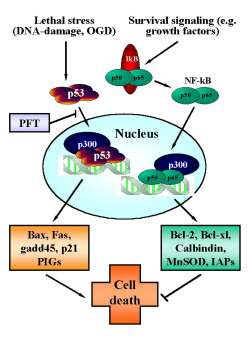
Figure 3. Reciprocal regulation of p53 and NF-kB transcriptional activity.
{kind=link}
In neurons, p53 is activated upon various (apoptotic) stress signals in neurons induced by DNA damage and oxygen glucose deprivation (OGD). Upon activation p53 translocates to the nucleus and initiates the transcription of proapoptotic factors including bax, gadd45, p21/Waf and other proapoptotic p53-inducible genes (PIGs). By contrast, constitutive survival signaling involves activation and nuclear translocation of NF-kB and induces synthesis of anti-apoptotic proteins such as bcl-2, bcl-xl, MnSOD, calbindin or inhibitors of apoptosis proteins (IAPs). For sufficient binding to the respective promotor sequences both transcription factors require co-transcription factors such as p300. Since the pool of p300 in the nucleus appears to be limited, the transcription factors p53 and NF-kB compete for binding to these cofactors to induce pro- or anti-apoptotic signaling. Thus, inhibition of p53 nuclear translocation by pifithrin-a (PFT) and its derivates indirectly promote NF-kB survival signaling, thereby further enhancing the anti-apoptotic effect of p53-inhibitors.
More recently, the importance of cytochrome c release and caspase activation in neuronal apoptosis has been supplemented by novel insights into a causal role of caspase-independent death signaling, such as the mitochondrial release of apoptosis inducing factor (AIF), in delayed neuronal death after cerebral ischemia. AIF is a 67 kDa NADH oxidase flavoprotein located in the mitochondrial intermembrane space with essential functions for optimal oxidative phosphorylation and efficient antioxidant defense. In many models of apoptosis, AIF is released from mitochondria and translocates to the nucleus where it induces chromatin condensation and large-scale (>50 kbp) DNA fragmentation in a variety of different cell types, including neurons. After oxygen glucose deprivation in vitro or after an ischemic insult in vivo AIF rapidly translocated to the nucleus of injured neurons and colocalized with DNA damage and apoptotic nuclear condensation. Of note, mitochondrial release of AIF occurred several hours before cytochrom c release and caspase-3 activation, suggesting that AIF is in the first line of cell death signaling after ischemia. On the other hand, the reduction of AIF protein levels in siRNA-treated cultured neurons or in mice carrying the harlequin (Hq) AIF gene mutation resulted in a significant reduction of neuronal cell death in the respective experimental models of ischemia by approximately 50%. These results expose AIF as a promising target for neuroprotective strategies in stroke therapy.
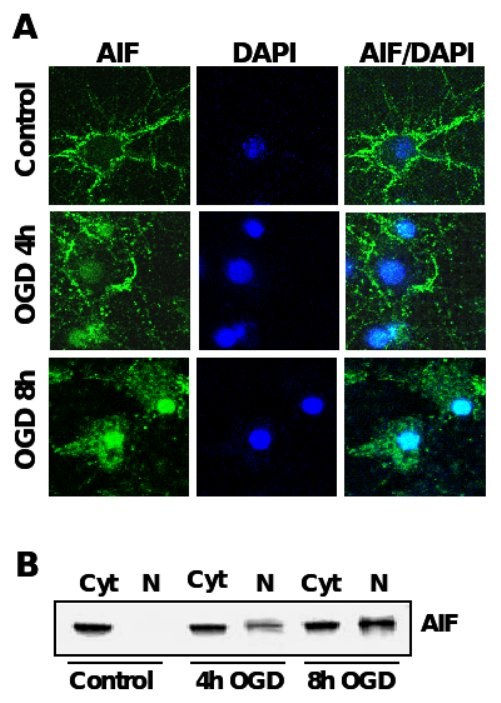
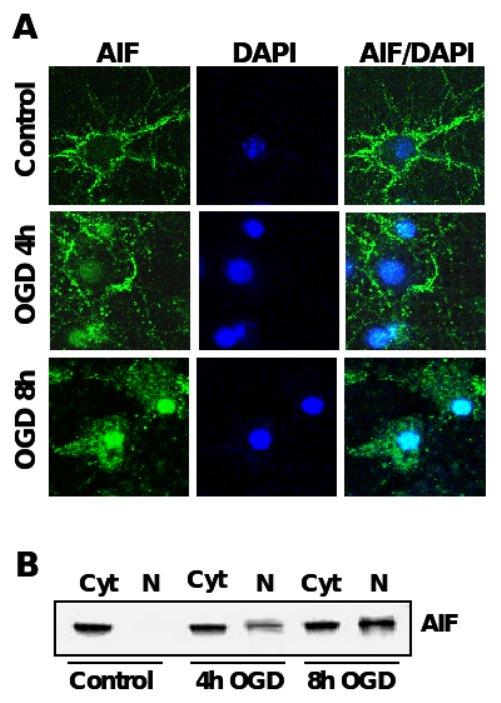
Figure 4. AIF-translocation precedes neuronal apoptosis after OGD.
{kind=link}
A. Immunocytochemistry of AIF (green) after oxygen glucose deprivation in primary rat neurons. Nuclei were counterstained with the DNA-binding dye DAPI (dark blue). B. AIF-Immunoblot analysis of protein extracts from cytosolic (Cyt) and nuclear extracts (N) of primary rat neurons exposed to OGD.
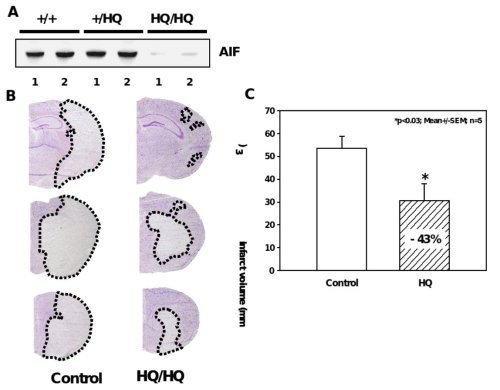
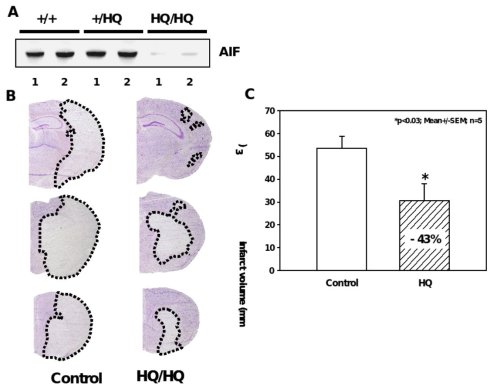
Figure 5. Significant reduction of ischemic brain damage in AIF-low expressing mice.
{kind=link}
A, Western blot analysis shows markedly reduced AIF protein levels in the brains of Hq/Hq mice as compared to homozygote wild type (+/+) and heterozygote (+/Hq) littermates. B, Nissl-stained brain sections of hippocampal (h) and striatal (s) brain regions obtained 24 h after reperfusion demonstrate significant reduction of infarcted brain tissue in Harlequin mice as compared to littermate controls. C, Infarct areas were evaluated histomorphometrically on 11 consecutive Nissl-stained brain sections (500 µm apart) throughout the infarct. The infarct volume was calculated on the basis of the histomorphometric data from the individual sections, showing a 43% reduction of the mean infarct volume in Hq mice as compared to wild type littermates.
Mitochondrial membrane permeabilization and subsequent release of apoptotic factors are key mechanisms during neuronal cell death associated with neurodegenerative diseases. Bcl-2 family proteins, e.g. the proapoptotic Bid, Bax or Bad and the anti-apoptotic Bcl-2, Bcl-xL, play a crucial role in the regulation of this mitochondrial checkpoint in neurons. In particular, cleavage of cytosolic Bid and subsequent mitochondrial translocation has been detected in many paradigms of neuronal cell death related to acute or chronic neurodegeneration.
Our recent results exposed the emerging role of Bid as an integrating key regulator of the intrinsic death pathway that amplifies caspase-dependent and caspase-independent execution of neuronal apoptosis. Therefore, pharmacological inhibition of Bid provides a promising therapeutic strategy in neurological diseases where programmed cell death is prominent.
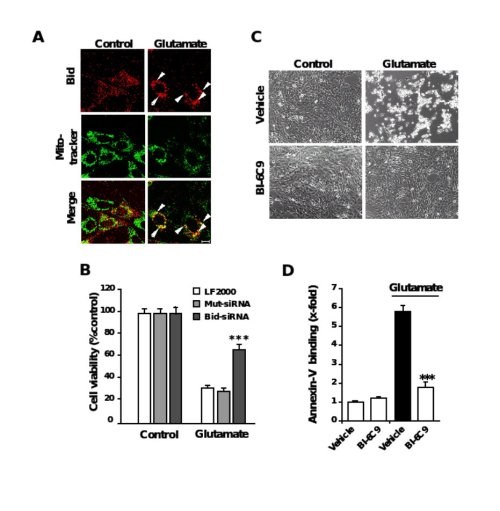
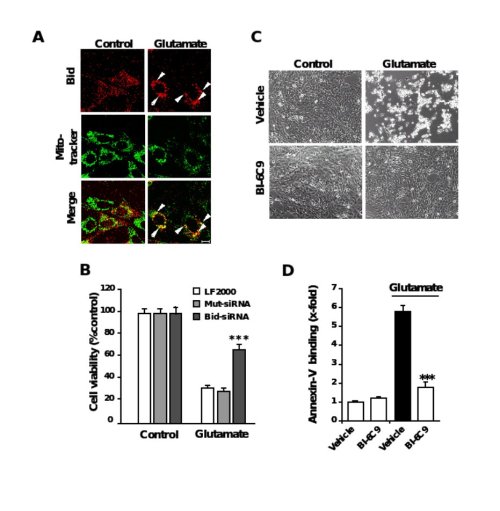
Figure 6. Role of Bid in neuronal apoptosis
{kind=link}
A, Fluorescence photomicrographs of HT-22 neurons expressing a Bid-DsRed fusion protein show co-localization of MitoTracker Green- and DsRed-signal in glutamate-damaged (5 mM, 5 h) cells. Note that the homogenous distribution of Bid in control cells significantly changes after glutamate-damage where Bid accumulates at mitochondria (yellow in merged panels). B, Bid-siRNA significantly attenuated glutamate-induced cell death as determined by MTT assay. C, Bid inhibitor BI-6C9 protects HT-22 cells against glutamate-induced apoptosis. Photomicrographs (10x objective) show morphological evidence for severe damage of HT-22 cells 17 h after glutamate (3 mM) exposure. Glutamate-treated cells lose their spindle-like morphology, shrink and detach from the culture well bottom; in contrast, cells pretreated with the Bid inhibitor BI-6C9 (10 µM) are fully protected against glutamate-induced death and are not different from the controls. D, FACS analysis of HT-22 cells after FITC- Annexin-V labeling to detect apoptotic cells. Exposure to glutamate (3 mM, 17 h) resulted in enhanced Annexin-V binding of apoptotic HT-22 cells compared to controls. BI-6C9 (10 µM, 1 h prior to damage) significantly reduced glutamate-induced apoptosis.