Hauptinhalt
Idiopathische Achalasie
Das internationale Forschungsnetzwerk arc (achalasia risk consortium) hat die Aufklärung der genetischen und zellbiologischen Ursachen der Achalasie zum Ziel. Die molekulargenetischen Analysen von arc werden am Zentrum für Humangenetik in Marburg durchgeführt. Zur Webpage von arc gelangen sie unter www.achalasie-konsortium.de.
Klinik der idiopathischen Achalasie
Bei der idiopathischen Achalasie handelt es sich um eine Motilitätsstörung der Speiseröhre. Sie ist durch eine Aperistaltik und die zunehmend eingeschränkte Erschlaffung des unteren Ösophagussphinkters (UÖS) gekennzeichnet. Hierdurch kann die aufgenommene Nahrung nicht mehr in den Magen transportiert werden. Ursächlich hierfür ist der Verlust von inhibitorischen Neuronen im Plexus myentericus, der die Muskelperistaltik der Speiseröhre und die Öffnung des UÖS steuert. Auch wenn die genauen Ursachen der Neurodegeneration noch nicht bekannt sind, scheinen autoimmune Prozesse bei Personen mit genetischer Disposition ursächlich zu sein (Gockel et al. (2010) Hum Genet). Die idiopathische Achalasie hat eine Lebenszeitprävalenz von 1:10.0000 und kommt gleichhäufig in beiden Geschlechtern vor (Gockel et al. (2012) Dtsch Arztebl Int).
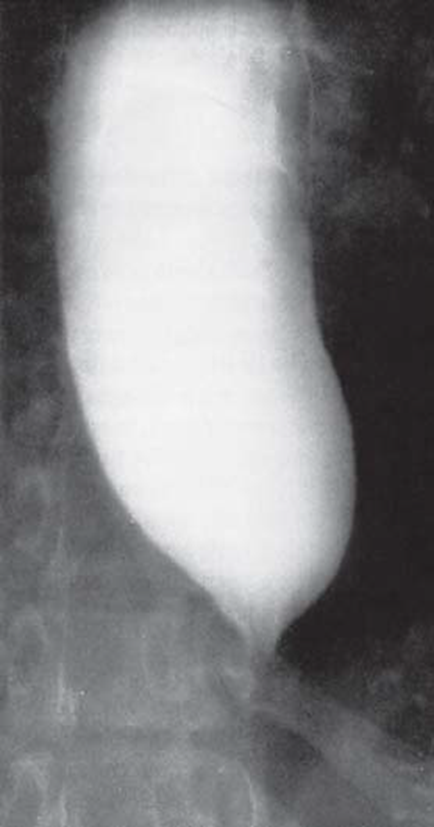
Klinisch äußert sich die idiopathische Achalasie durch Schluckstörungen, Regurgitation von Nahrung, retrosternalen Schmerzen und Gewichtsverlust. Durch den eingeschränkten Nahrungstransport stellt sich die Speiseröhre als massiv dilatierter Megaösophagus dar (s. Abbildung 1).
Die aufgestaute Nahrung kann bei Übertritt in die Luftröhre zu bronchopulmonalen Infektionen und Aspirationspneumonien führen. Durch die chronische Mikroaspiration von Nahrung zeigen 50% der Patienten eine eingeschränkte Lungenfunktion (Gockel et al. (2012) Dtsch Arztebl Int). Infolge der chemischen und mechanischen Schleimhautirritation führt die Nahrungsretention auch zu Entzündungen der Speiseröhre. In deren Folge entwickeln 4-6% der Patienten ein Plattenepithelkarzinom (Gockel et al. (2012) Dtsch Arztebl Int). Als Behandlungsmöglichkeiten stehen die chirurgische Spaltung des UÖS (u. a. sog. Heller-Kardiomyotomie) und die pneumatische Dilatation der Speiseröhre zur Verfügung.
Genetik der idiopathischen Achalasie
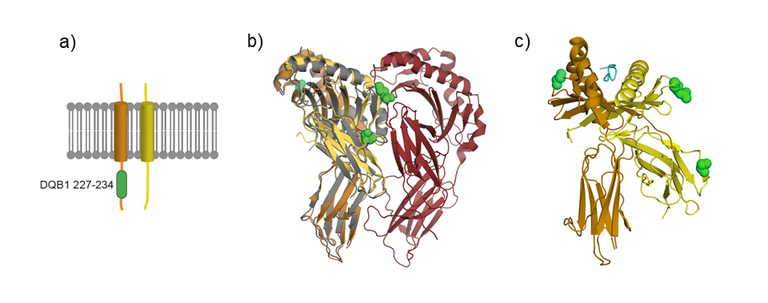
Bei der idiopathischen Achalasie handelt es sich um eine multifaktorielle Erkrankung (Gockel et al. (2010) Hum Genet). Mit dem Ziel, genetische Risikovarianten für die Achalasie zu identifizieren, führten wir die erste systematische Assoziationsanalyse an einem großen europäischen Fall-Kontrollkollektiv durch (> 1.000 Patienten, > 2.200 Kontrollen) (Gockel et al. (2014) Nat Genet). In dem Kollektiv testeten wir > 190.000 häufige genetische Varianten auf Assoziation zur Krankheit. Die am stärksten Achalasie-assoziierte Variante war rs28688207 (P = 1,73 x 10-19) und liegt in der sog. HLA-Region auf Chromosom 6. Das Risikoallel zeigt eine Frequenz von 9,9% bei Patienten und 4,7% bei Kontrollen (Relatives Risiko (RR) = 2,43). Funktionell führt die identifizierte Variante zu einem alternativ gespleißten Exon im Gen HLA-DQB1 und auf Proteinebene zu einer Insertion von 8 Aminosäuren, die im zytoplasmatischen Teil von HLA-DQβ1 lokalisiert ist. Die Assoziation zwischen der Insertion bzw. der genetischen Risikovariante und der Achalasie konnten wir in einem unabhängigem europäischem Fall-Kontrollkollektiv bestätigen (> 400 Patienten, > 1.000 Kontrollen, 9,3% bei Patienten, 3,8% bei Kontrollen, P = 7,09 x 10-09, RR = 2,59). Anschließend testeten wir, ob unabhängige Achalasie-Risikovarianten in der HLA-Region auf Chromosom 6 lokalisiert sind. Hierbei zeigte sich eine genomweit-signifikante Achalasie-Assoziation für eine genetische Variante, die auf Proteinebene zu einem Aminosäureaustausch an Positionen 41 von HLA-DQα1 führt. An der Position kommen die Aminosäuren Lysin (K) oder Arginin (R) vor, wobei erstere krankheitsdisponierend ist (12,5% bei Patienten, 7,1% bei Kontrollen, P = 2,37 x 10-12, RR = 1,86). Folgend testeten wir, ob weitere Risikovarianten in der HLA-Region auf Chromosom 6 lokalisiert sind. Hierdurch konnten wir eine dritte genomweit-signifikante Achalasie-Assoziation für eine genetische Variante finden, die auf Proteinebene zu einem Aminosäureaustauch an Position 45 von HLA-DQβ1 führt. An dieser Position kommen die Aminosäuren Glutaminsäure (E) und Glycin (G) vor. Erstere stellt das Achalasie-Risikoallel dar (24,9% bei Patienten, 22,4% bei Kontrollen, 1,20 x 10-09, RR = 1,47).
Auf der Proteinebene bilden HLA-DQβ und HLA-DQα als Heterodimer den HLA-DQ Komplex, der zu den HLA Klasse-II Molekülen zählt und auf Antigen-präsentierenden Zellen (APC) exprimiert wird. HLA Klasse-II Moleküle präsentieren CD4+ T-Zellen Antigene, die extrazellulär mittels Phagozytose oder Pinozytose aufgenommen werden, und lösen damit eine Immunantwort des Organismus aus. Unsere Forschungsdaten legen nahe, dass der genetischen Variabilität im HLA-DQ Komplex und damit der Immunantwort ursächliche Bedeutung am Entstehungsprozess der Achalasie zukommt.
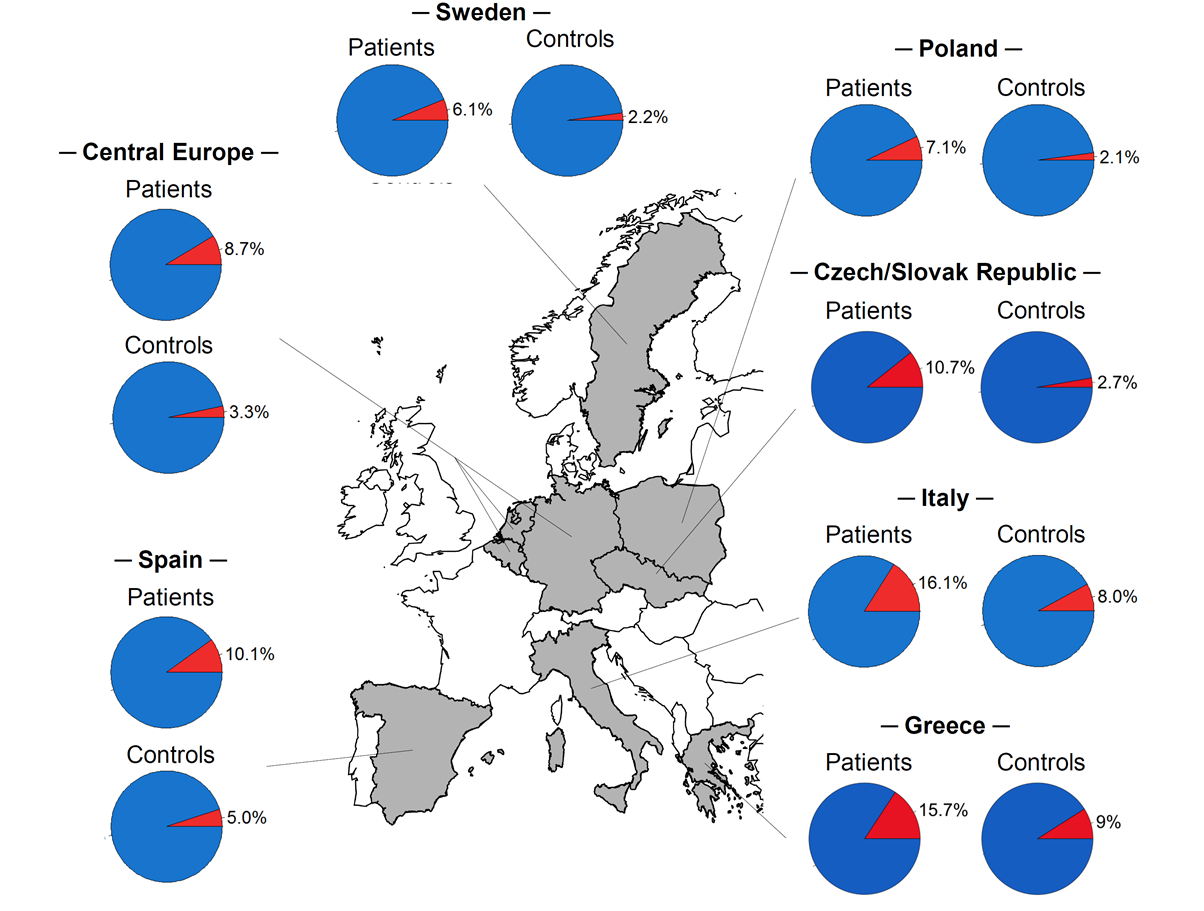
In weiterführenden Untersuchungen gingen wir der Frage nach, ob die stärkste Achalasie-assoziierte Risikovariante (rs28688207), die in der HLA-Region auf Chromosom 6 lokalisiert ist und auf Proteinebene zur 8-Aminosäureinsertion in HLA-DQβ1 führt, gleich häufig in europäischen Subpopulationen vorkommt (Becker et al. (2016) Eur J Hum Genet). Dabei konnten wir einen geografischen Nord-Süd-Gradienten der Insertion mit höherer Frequenz in südeuropäischen Populationen (Italien, Spanien) gegenüber mittel- und nordeuropäischen Populationen nachweisen (Belgien, Deutschland, Niederlande, Polen, Schweden). Diesen Nord-Süd-Gradienten konnten wir in der Zwischenzeit durch Hinzuziehen von Fall-Kontrollkollektiven aus der Tschechischen und Slowakischen Republik sowie Griechenland bestätigten (Vackova et al. (2018) UEG Journal) (s. Abbildung 3). Die Ursache für den geografischen Nord-Süd-Gradienten mit höherer Frequenz der Insertion in Südeuropa ist derzeit nicht bekannt. Vermutlich werden hieran evolutionäre Mechanismen beteiligt sein, wie genetischer Drift oder natürliche Selektion im Kontext zu unterschiedlichen Umwelteinflüssen. Bei letztgenannter Untersuchung zeigte sich überdies, dass die 8-Aminosäureinsertion in HLA-DQβ1 häufiger einer bestimmten Unterform der idiopathischen Achalasie zugrunde liegt, bei der die Neurodegeneration der inhibitorischen Neurone im Plexus myentericus nach der Chicago Classification (CC) basierend auf den Ergebnissen der High-Resolution Manometry (HRM) besonders ausgeprägt ist.
Ansprechpartner
Prof. Dr. Johannes Schumacher
Zur Webpage von arc gelangen sie unter www.achalasie-konsortium.de.
Ausgewählte Publikationen
Vackova Z, Niebisch S, Triantafyllou T, Becker B, Hess T, Kreuser N, Kanoni S, Deloukas P, Schüller V, Heinrichs SKM, Thieme R, Nöthen MM, Knapp M, Spicak J, Gockel I, Schumacher J, Theodorou D, Martinek J. First genotype-phenotype study reveals HLA-DQB1 insertion heterogeneity in high-resolution manometry achalasia subtypes. United European Gastroenterology J. 2019 7:45–51.
Becker J, Niebisch S, Ricchiuto A, Schaich EJ, Lehmann G, Waltgenbach T, Schafft A, Hess T, Lenze F, Venerito M, Hüneburg R, Lingohr P, Matthaei H, Seewald S, Scheuermann U, Kreuser N, Veits L, Wouters MM, Gockel HR, Lang H, Vieth M, Müller M, Eckardt AJ, von Rahden BH, Knapp M, Boeckxstaens GE, Fimmers R, Nöthen MM, Schulz HG, Gockel I, Schumacher J. Comprehensive epidemiological and genotype-phenotype analyses in a large European sample with idiopathic achalasia. Eur J Gastroenterol Hepatol. 2016 28:689-95.
Becker J, Haas SL, Mokrowiecka A, Wasielica-Berger J, Ateeb Z, Bister J, Elbe P, Kowalski M, Gawron-Kiszka M, Majewski M, Mulak A, Janiak M, Wouters MM, Schwämmle T, Hess T, Veits L, Niebisch S, Santiago JL, de León AR, de la Serna JP, Urcelay E, Annese V, Latiano A, Fumagalli U, Rosati R, Laghi L, Cuomo R, Lenze F, Sarnelli G, Müller M, von Rahden BH, Wijmenga C, Lang H, Czene K, Hall P, de Bakker PI, Vieth M, Nöthen MM, Schulz HG, Adrych K, Gąsiorowska A, Paradowski L, Wallner G, Boeckxstaens GE, Gockel I, Hartleb M, Kostic S, Dziurkowska-Marek A, Lindblad M, Nilsson M, Knapp M, Thorell A, Marek T, Dąbrowski A, Małecka-Panas E, Schumacher J. The HLA-DQβ1 insertion is a strong achalasia risk factor and displays a geospatial north-south gradient among Europeans. Eur J Hum Genet. 2016 24:1228-31. doi: 10.1038/ejhg.2015.262.
Gockel I, Becker J, Wouters MM, Niebisch S, Gockel HR, Hess T, Ramonet D, Zimmermann J, Vigo AG, Trynka G, de León AR, de la Serna JP, Urcelay E, Kumar V, Franke L, Westra HJ, Drescher D, Kneist W, Marquardt JU, Galle PR, Mattheisen M, Annese V, Latiano A, Fumagalli U, Laghi L, Cuomo R, Sarnelli G, Müller M, Eckardt AJ, Tack J, Hoffmann P, Herms S, Mangold E, Heilmann S, Kiesslich R, von Rahden BH, Allescher HD, Schulz HG, Wijmenga C, Heneka MT, Lang H, Hopfner KP, Nöthen MM, Boeckxstaens GE, de Bakker PI, Knapp M, Schumacher J. Common variants in the HLA-DQ region confer susceptibility to idiopathic achalasia. Nat Genet. 2014 46:901-4. doi: 10.1038/ng.3029.
Wouters MM, Lambrechts D, Becker J, Cleynen I, Tack J, Vigo AG, Ruiz de León A, Urcelay E, Pérez de la Serna J, Rohof W, Annese V, Latiano A, Palmieri O, Mattheisen M, Mueller M, Lang H, Fumagalli U, Laghi L, Zaninotto G, Cuomo R, Sarnelli G, Nöthen MM, Vermeire S, Knapp M, Gockel I, Schumacher J, Boeckxstaens GE. Genetic variation in the lymphotoxin-α (LTA)/tumour necrosis factor-α (TNFα) locus as a risk factor for idiopathic achalasia. Gut. 2014 63:1401-9.
Gockel I, Müller M, Schumacher J. Achalasia--a disease of unknown cause that is often diagnosed too late. Dtsch Arztebl Int. 2012 109:209-14.
Gockel HR, Gockel I, Schimanski CC, Schier F, Schumacher J, Nöthen MM, Lang H, Müller M, Eckardt AJ, Eckardt VF. Etiopathological aspects of achalasia: lessons learned with Hirschsprung's disease. Dis Esophagus. 2012 25:566-72.
Gockel HR, Schumacher J, Gockel I, Lang H, Haaf T, Nöthen MM. Achalasia: will genetic studies provide insights? Hum Genet. 2010 128: 353-364.