Main Content
Moleculare Dynamic (MD) Simulation
The idea of an MD simulation is to numerically integrate Newton's classic equations of motion, in order to construct the positions of a molecule changing with time (trajectories). Thereby all involved atoms have to be considered, especially those in the environment. Mostly the particles to be observed are on a surface or in solution. Therefore the number of particles to consider is infinitely large to first-order approximation.
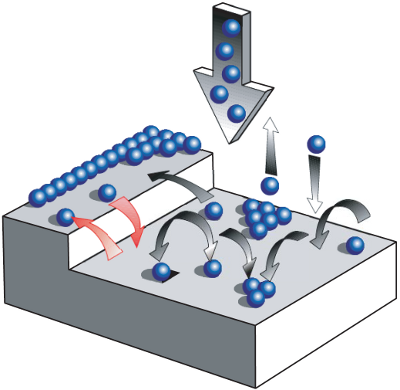
Figure 1: Numerous processes are relevant to thin film growth (from phys. stat. sol. (a) 201 (2004) Schreiber). Besides movements over step edges and nucleation especially the diffusion on the substrate surface is crucial.
As the interaction with the environment normally takes place on a significantly faster time scale than the motion of the observed molecule itself, the effects of the environment are summarized as noise affecting the molecule. The result is the Langevin equation. This equation describes how the molecule converts the thermal energy of the environment into kinetic energy (fluctuation) and how it releases kinetic energy to the environment (dissipation). Both fluctuation and dissipation depend on temperature, mass and friction. This is specified by the fluctuation-dissipation theorem.
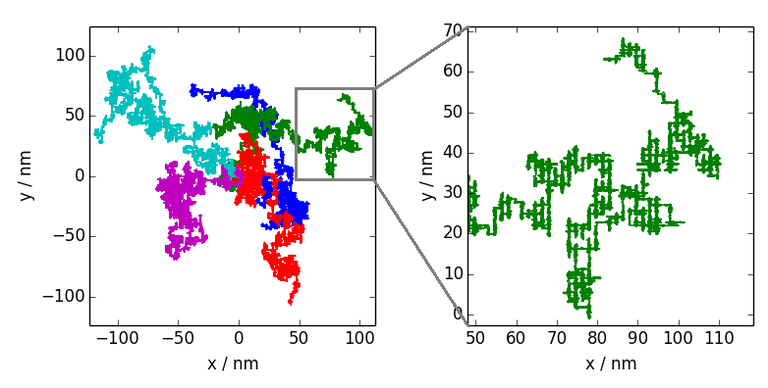
Figure 2: Depicted are five trajectories of a molecule moving over the surface. What seems to be normal Brownian motion at first sight reveals an underlying structure on closer inspection: The motion mainly occurs on a rectangular lattice.
Besides the explained thermally fluctuating forces the Langevin equation contains potentials for interactions between the diffusing particles themselves and the surface. This interaction strongly depends on system structure and usually calculations are very expensive. The approaches vary from educated guessing to pair potentials (molecular mechanics) to quantum mechanical (DFT) measurements.
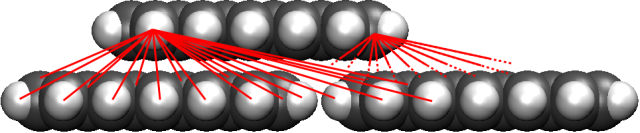
Figure 3a: Interaction of pentacene with a pentacene surface. The interaction potential is calculated by pairwise summation of the interactions between all carbon-hydrogen combinations.
Figure 3b: Pentacene diffuses on a pentacene surface at room temperature. The pentacene molecules forming the surface are chemically bound to a copper surface. The trajectory, depicted in 5 seconds, corresponds to a real time of 0.5 nanoseconds. Thus the animation is slowed down 1010-fold.
Typical questions are comparisons with theoretical predictions or experiments, which can be calculated from the trajectories.
Figure 4: Plotting the square distance vs time a linear relation is obtained with the diffusion coefficient as proportionality constant. The top curve is averaged over the five trajectories shown above, whereas for the bottom diagram 300 trajectories were calculated and averaged. This shows the correlation more clearly.
To perform extensive simulations our research group has a customized high performance computer available.
A publication from our group utilising molecular dynamics simulations:
- Coupling between diffusion and orientation of pentacene molecules on an organic surface.
Paul Rotter, Barbara A. J. Lechner, Antonia Morherr, David M. Chisnall, David J. Ward, Andrew P. Jardine, John Ellis, William Allison, Bruno Eckhardt, Gregor Witte
Nature Materials 15 (4), 397-400 (2016) • DOI: 10.1038/nmat4575
Full Text