Main Content
2018
ADV. MATER. INTERFACES: "Interface Structure and Evolution of DNTT Films on Noble Metal Substrates."
Maximilian Dreher, Daniel Bischof, Felix Widdascheck, Andrea Huttner, Tobias Breuer and Gregor Witte
Adv. Mater. Interfaces 5 (21), 1800920 (2018) • DOI: 10.1002/admi.201800920
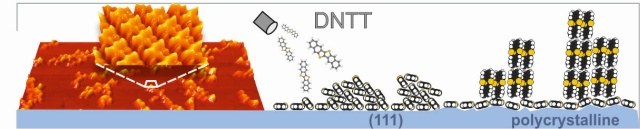
Dinaphthothienothiophene (DNTT) is a promising new organic semiconductor which combines high charge-carrier mobility with chemical robustness. Although the properties of organic electronic devices are largely determined by the interfaces with electrodes, the interface between DNTT and metals is hardly studied so far. Here, we have examined the interface-structure of DNTT on Ag(111) and multilayers and compared them with films grown on polycrystalline and (111) substrates of silver and gold. In the seed-layer regime, we identify two different interface-structures formed by exclusively flat-lying molecules and a herringbone-arrangement for increased coverages. Combining LEED, STM, and NEXAFS measurements, we have derived precise structure data for both phases and compare their respective adsorption energies derived from thermal-desorption-spectroscopy measurements to explain why both phases are formed. On all examined surfaces DNTT multilayers consist of discrete fibers reflecting a notable dewetting but exhibit different molecular orientations: Molecules are recumbently arranged and form (12-1) oriented islands on the (111) metal substrates, but uprightly oriented on polycrystalline noble metal surfaces. Complementary work-function measurements yield rather similar values for DNTT monolayers on single- and polycrystalline metal substrates, while distinct differences are found for thicker films. Our results appear important for improved transistor geometries, in particular the realization of vertical field-effect-transistors.PHYS. CHEM. CHEM. PHYS.: "Polarized absorbance and Davydov splitting in bulk and thin-film pentacene polymorphs."
C. Cocchi, T. Breuer, G. Witte, C. Draxl
Phys. Chem. Chem. Phys. 20, 29724-29736 (2018) • DOI: 10.1039/C8CP06384B
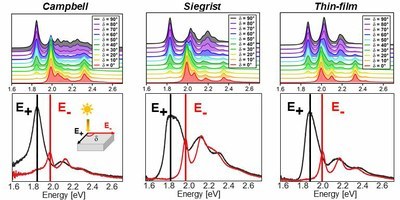
Pentacene is one of the most studied organic materials and in particular its optical properties have been the subject of an intense research during the last two decades. In spite of such a widespread interest and of the extensive knowledge achieved so far, a number of issues are still debated. One of the most relevant questions concerns the role of polymorphism and how it affects the lowest-energy exciton, which appears in the visible region and is subject to a sizable Davydov splitting. We address this problem in a combined theoretical and experimental work, where the optical absorption properties of three pentacene polymorphs are investigated within the whole energy range of visible light. Optical spectra computed from first principles in the framework of many-body perturbation theory are directly compared with the polarization-resolved absorbance, measured for three different pentacene phases (the two bulk polymorphs and the thin-film phase). In this way, we unambiguously identify the two Davydov components of the first exciton and the optical fingerprints of each considered phase. With very good agreement between theory and experiment, we show that all polymorphs exhibit common features at the absorption onset, while phase-dependent characteristics appear only above 2 eV. We discuss the character of the lowest-lying singlet and triplet excitons, including dark ones, highlighting the contributions from the electronic bands and the role of the electron-hole interaction and of the local-field effects.J. PHYS. CHEM. C: "Assignment of NEXAFS Resonances in Alkanethiols and Their Implication on the Determination of Molecular Orientation of Aliphatic SAMs."
Johannes Völkner, Michael Klues and Gregor Witte
J. Phys. Chem. C 122 (29), 16810-16820 (2018) • DOI: 10.1021/acs.jpcc.8b04342
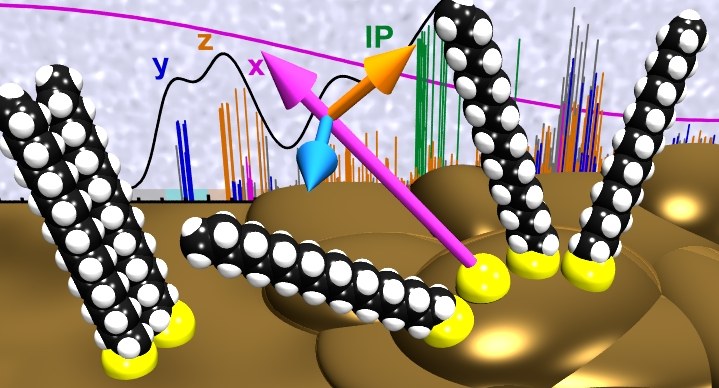
Near-edge X-ray absorption fine structure (NEXAFS) spectroscopy has become a widely used technique to analyze the molecular orientation of functional adlayers by analyzing the dichroism of NEXAFS resonances. In contrast to π-conjugated systems, which exhibit distinct π-resonances whose transition dipole moments are oriented perpendicular to the aromatic planes, the analysis of purely aliphatic systems is not as straightforward and requires a precise identification of the underlying resonances and their polarization. Here we analyze the carbon K-edge NEXAFS signature and its dichroism of a self-assembling monolayer (SAM) formed by octadecanethiol (ODT) on gold representing the widely studied class of alkanethiol-SAMs. Employing density functional theory calculations and using the transition potential method, the polarizations of all NEXAFS resonances at excitation energies around and below the ionization threshold are precisely determined. This information is then used to simulate expected dichroism curves for various adlayer arrangements which are compared with our experimental findings for ODT-SAMs on single crystalline Au(111) and polycrystalline gold surfaces. This analysis demonstrates a novel strategy to precisely deduce the molecular orientation from the experimental NEXAFS data of aliphatic SAMs.PHYS. STAT. SOL. A: "Structure and thermal stability of stilbenedithiol SAMs on Au(111)."
Johannes Völkner, Asif Bashir, Wolfgang J. Parak and Gregor Witte
Phys. Stat. Sol. A 215 (15), 1700859 (2018) • DOI: 10.1002/pssa.201700859
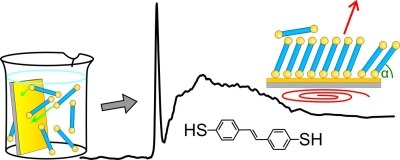
Self-assembling monolayers (SAMs) of dithiols play an important role as divalent linker layer in applications such as photoelectrochemical sensors. However, their formation processes and film structure are not fully understood yet. Herein, we investigate a SAM of aromatic transstilbenedithiol (StDT) on Au(111) surfaces. Using complementary characterization techniques we investigated the dependence of the film quality on preparation parameters, such as used solvent, temperature and ambient conditions during and subsequent to preparation. Furthermore, we found a remarkably high thermal stability which we ascribe to disulfide bridging between the endgroups of the individual molecules.J. PHYS. CHEM. C: "Copper Phthalocyanine as Contact Layers for Pentacene Films Grown on Coinage Metals."
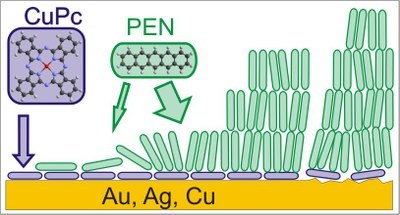
Alexander Mänz, Alrun Aline Hauke and Gregor Witte
J. Phys. Chem. C 122 (4), 2165-2172 (2018) • DOI: 10.1021/acs.jpcc.7b10324
The metal-semiconductor interface determines the efficiency of charge carrier injection into any organic electronics device. Control of this interface, its structure and morphology is therefore essential for device improvement. In this study, we analyze the approach of controlling semiconductor morphology at this interface by insertion of a copper phthalocyanine (CuPc) monolayer as a primer between Ag(111), Au(111) and Cu(100) surfaces and the organic semiconductor pentacene (PEN). Controlled monolayer formation is facilitated by thermal desorption of excess multilayers, monitored via thermal desorption spectroscopy (TDS), X-ray photoelectron spectroscopy (XPS) and scanning tunneling microscopy (STM), and the growth of PEN on the resultant monolayer primers is investigated by near-edge X-ray absorption spectroscopy (NEXAFS), atomic force microscopy (AFM) and STM. While well-ordered CuPc monolayers with flat-lying molecules are formed on Au(111) and Ag(111), no long-range order is observed on Cu(100). Subsequently deposited PEN molecules initially adopt a recumbent orientation with their long axis oriented parallel to the surface, while upon further deposition this structure is metastable as molecules adopt an upright orientation beyond the bilayer and form (001) oriented films. Although the recumbent orientation of the CuPc primer layer is not transferred to thicker PEN films, which is attributed to the geometrical inequality of the two molecules, a distinct dewetting, as found for PEN films grown on bare metal surfaces, is efficiently suppressed. This effect is reproducible even for polycrystalline Au surfaces, which resemble the situation of metal contacts in devices.PNAS: "Low-lying excited states in crystalline perylene."
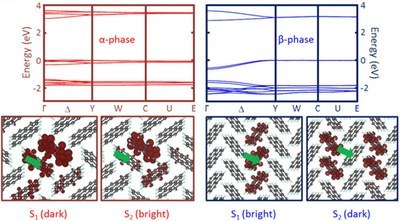
Tonatiuh Rangel, Andre Rinn, Sahar Sharifzadeh, Felipe H. da Jornada, André Pick, Steven G. Louie, Gregor Witte, Leeor
Kronik, Jeffrey B. Neaton, and Sangam Chatterjee
PNAS 115 (2), 284-289 (2018) • DOI: 10.1073/pnas.1711126115
Organic materials are promising candidates for advanced optoelectronics and are used in light-emitting diodes and photovoltaics. However, the underlying mechanisms allowing the formation of excited states responsible for device functionality, such as exciton generation and charge separation, are insufficiently understood. This is partly due to the wide range of existing crystalline polymorphs depending on sample preparation conditions. Here, we determine the linear optical response of thin-film single-crystal perylene samples of distinct polymorphs in transmission and reflection geometries. The sample quality allows for unprecedented high-resolution spectroscopy, which offers an ideal opportunity for judicious comparison between theory and experiment. Excellent agreement with first-principles calculations for the absorption based on the GW plus Bethe–Salpeter equation (GW-BSE) approach of many-body perturbation theory (MBPT) is obtained, from which a clear picture of the low-lying excitations in perylene emerges, including evidence of an exciton–polariton stopband, as well as an assessment of the commonly used Tamm–Dancoff approximation to the GW-BSE approach. Our findings on this well-controlled system can guide understanding and development of advanced molecular solids and functionalization for applications.CRYSTENGCOMM: "Crystalline Packing in Pentacene-like Organic Semiconductors."

M. Klues and G. Witte
CrystEngComm 20, 63-74 (2018) • DOI: 10.1039/C7CE01700F
Since optoelectronic properties of organic semicond uctors (OSCs) are largely affected by the molecular packing in the solid phase, further advances of such materials require c omprehensive structure-property interrelations beyo nd single molecule considerations. While single molecular ele ctronic properties can be tailored by synthetic mea ns and their electronic properties can be reliably predicted by quantum chemical calculations, crystal structure pr edictions of such van der Waals bond solids remain challenging. Here we a nalyze correlations between the molecular structure and the resulting packing motifs adopted in the crystalline phases of the prototypical OSC pentacene as well as various differently substituted but similarly shaped π -conjugated molecules. Based on a Hirshfeld surface analysis and related fingerprint plots, specific contact points and their distributi on are identified which allows a classification of different structure groups. Comparing the fingerprint plots with corresponding molecular properties such as electrostatic contour plots as well as quadrupole and polarizability tensors, which were c alculated by density functional theory, allows rati onalizing structure determining specific intermolecular interactions. O ur analysis shows in particular that molecules with uniform electrostatic potential at their periphery favor a herringbone pa cking, while the highly electronegative substituent s (O, N and F) enable the formation of H-bonds and prefer slip-stacking o r criss-cross packing motifs. The present correlati ons might be useful guidelines for future strategies to synthesis new OSCs.