Main Content
Applications
Quantum chemical applications in our group usually are guided by the ideas of collaboration partners. Selected examples:
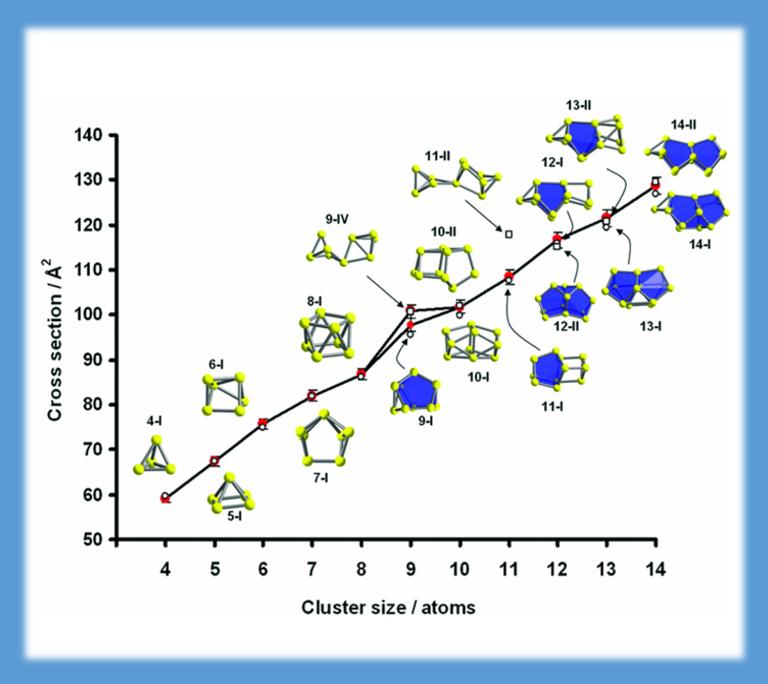
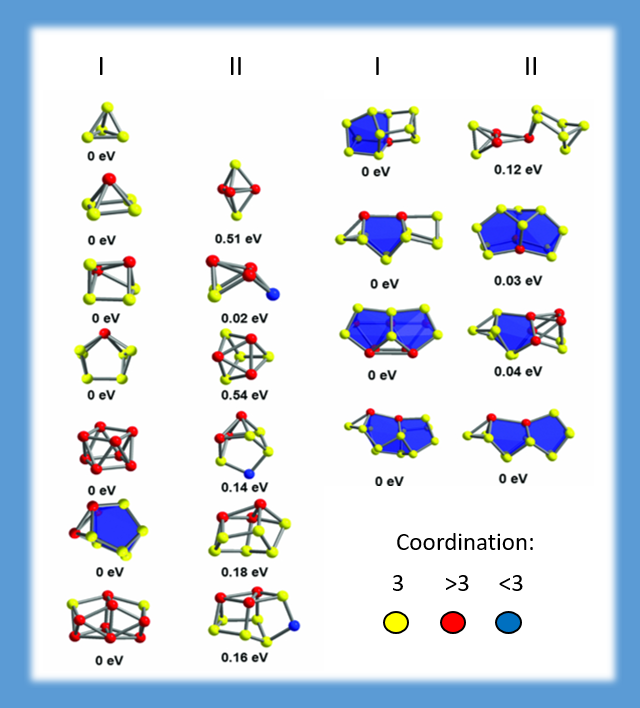
Beam experiments (group of M.M. Kappes, Karlsruhe Institute of Technology) yield mass-selected charged clusters of (semi-)metal atoms, for which the cross sections were determined by scattering in a helium drift cell. For the clarification of the geometric structure, we employed a density functional theory based genetic algorithm to determine the most favourable isomers for each size. For the present example of bismuth clusters it turned out that the two criteria - low energy and matching cross section - can be fulfilled simultaneously only when spin-orbit coupling is regarded in the DFT calculations (J. CHEM. PHYS. 136, 154309 (2012)).
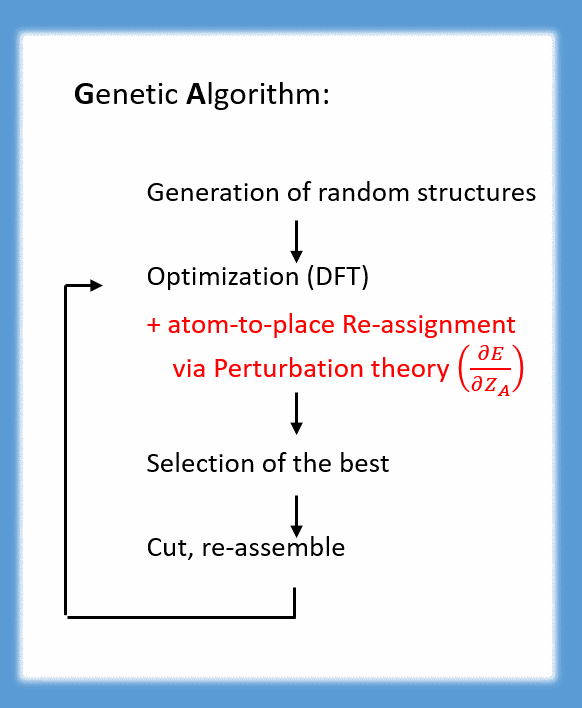
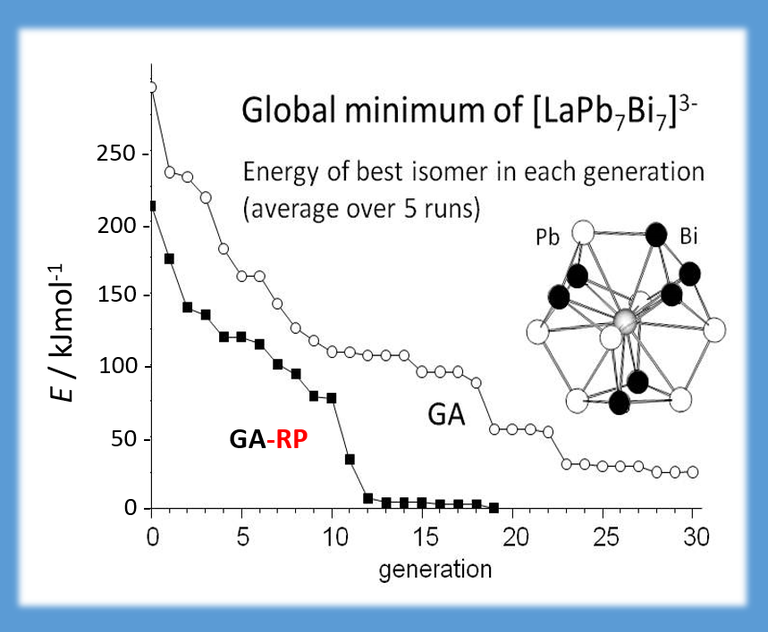
Binary (and ternary) clusters may be predicted with genetic algorithms, too. Due to the high number of possible distributions of atoms to positions, this means much more effort. Luckily, if the electronic structure is not too sensitive to changes in the nuclear charge (or equivalently: as long as the pseudo-element concept applies), efficiency of genetic algorithms can be increased very much when extending them by a first-order perturbation step in the nuclear charge (J. CHEM. PHYS. 141, 134103 (2014)). In this way, it was possible to predict the global minimum of e.g. [LaPb7Bi7]3- before the cluster was synthesized (CHEM. EUR. J. 21, 386-394 (2015)).
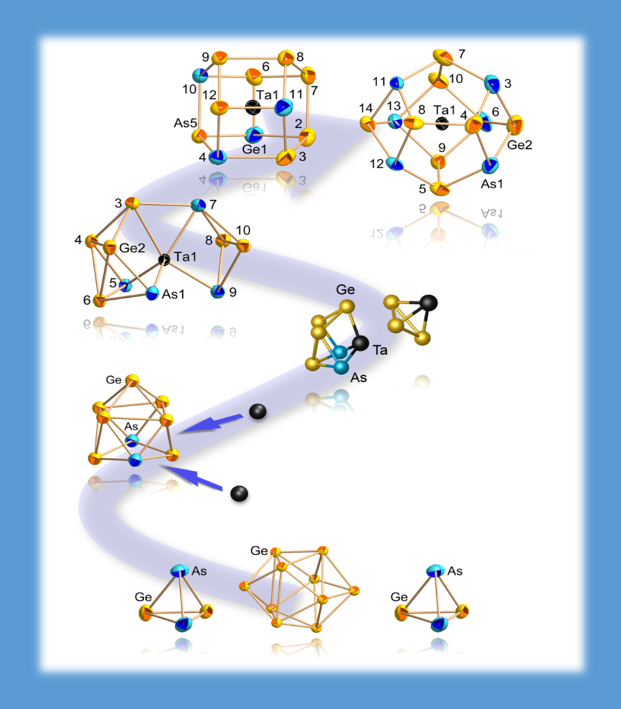
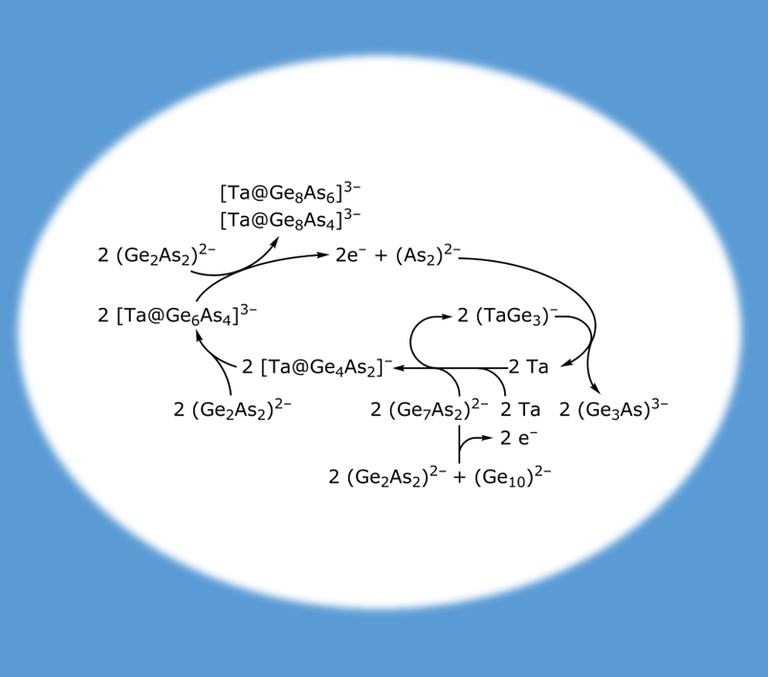
Shedding light on the formation of cluster species is not easy. In this example, an experimentally found intermediate and its confirmation by an extended genetic algorithm as well as an almost isoenergetic isomer found in the same procedure were the first step. Subsequently, proposed intermediate steps were investigated by quantum chemistry, including possible transformation pathways, in the end leading to a proposal for the overall formation cycle (NATURE COMMUNICATIONS 7, 10480 (2016)).
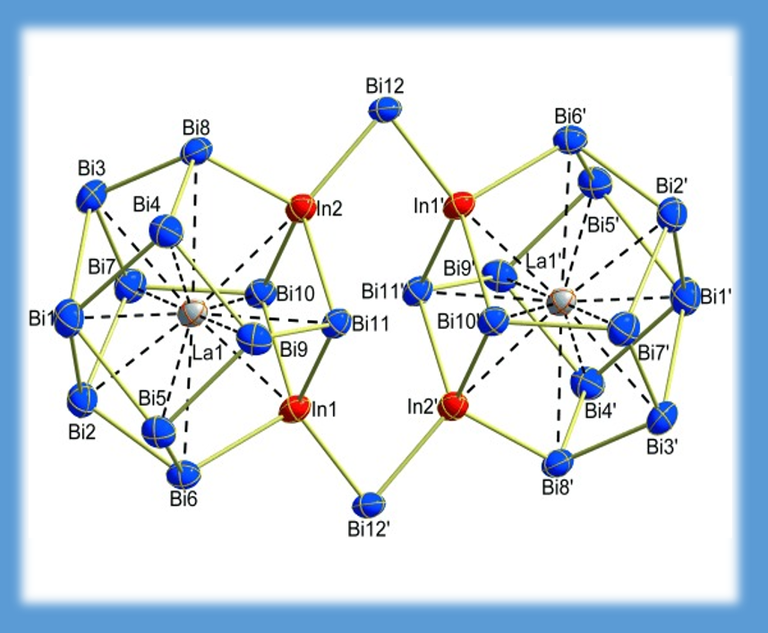

The two [La@In2Bi11] units in the ternary anion {[La@In2Bi11](μ-Bi)2[La@In2Bi11]}6- (left) are linked via two Bi atoms. It is almost a textbook example of MO theory: The four binding orbitals (right) consist of the two HOMOs of the fragments combined in symmetric/antisymmetric matter together with the matching p orbitals of the two Bi atoms (CHEM. EUR. J. 18, 13589-13595 (2012)).
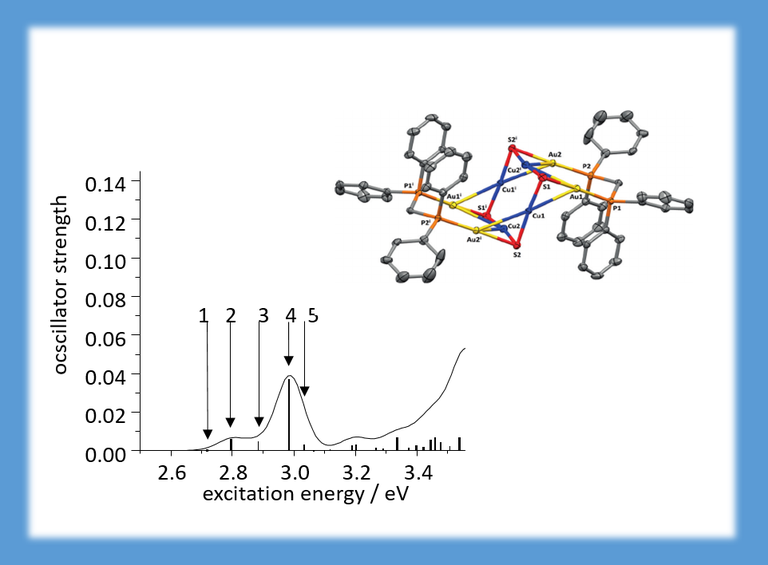
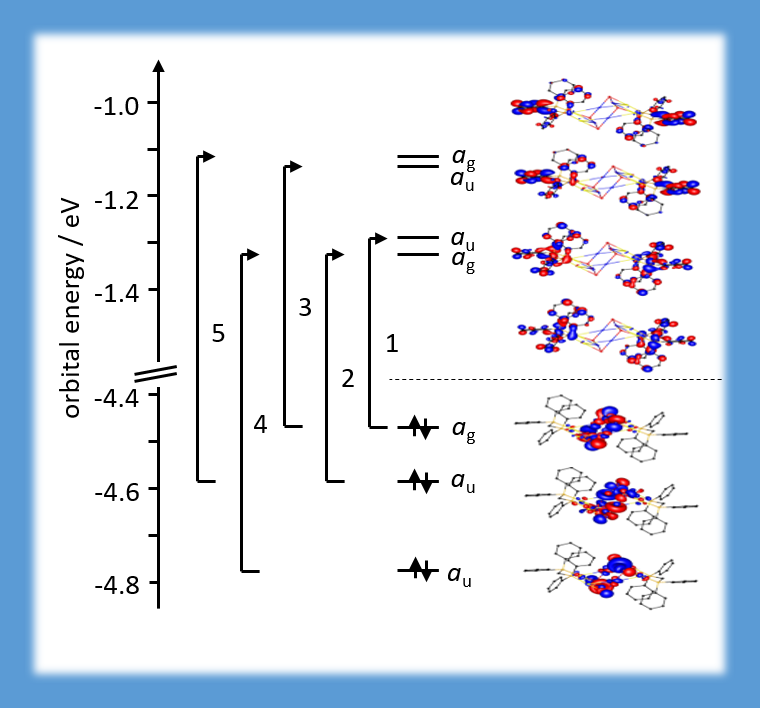
For this mixed Au-Cu cluster, photoluminescence properties were investigated with time-dependent density functional theory. The lowest energy band in the excitation energy spectrum (between 2.7 and 3.1 eV) consists of five excitations involving the three highest occupied and the four lowest unoccupied orbitals. The three highest occupied orbitals are mainly formed from the Cu4S4-ring in the centre, whereas the four lowest unoccupied orbitals are located at the phenyl ligands. The band of lowest excitations thus has strong metal-to-ligand (and inter-ligand) charge-transfer character (CHEM. EUR. J. 22, 18378–18382 (2016)).
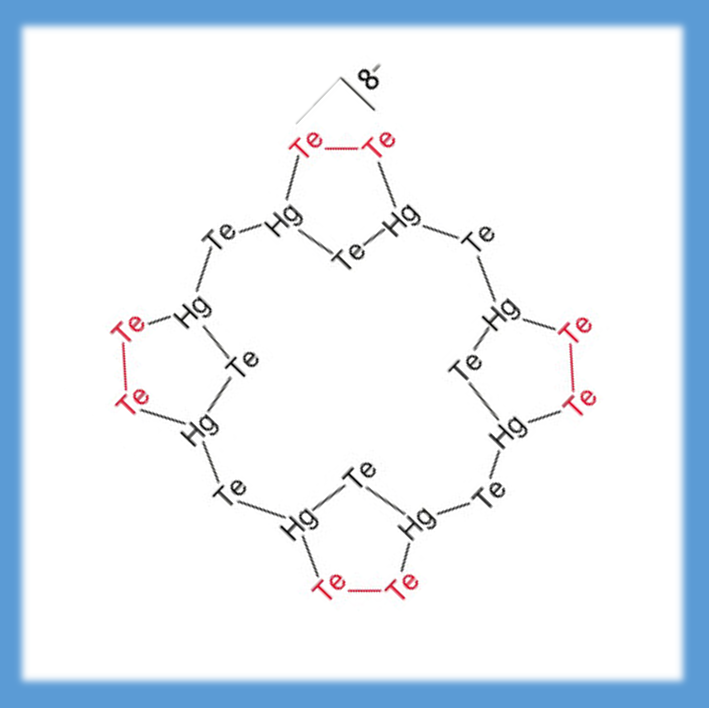
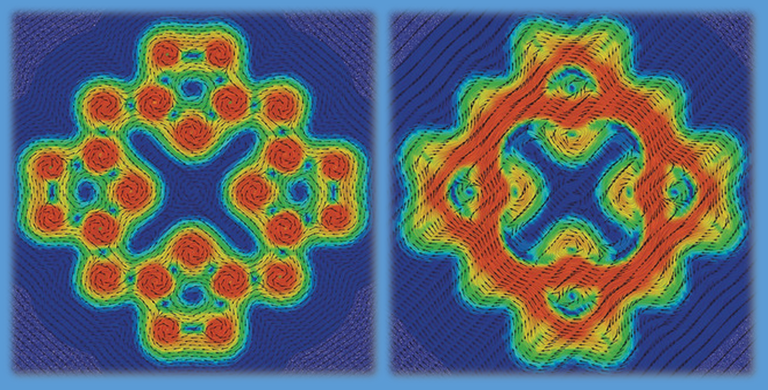
There are several possibilities to correlate the concept of aromaticity with numbers form quantum chemical treatments. Most elegant is the calculation of magnetically induced ring currents, obtained from the magnetic response by employing the law of Biot-Savart. The compound (left) looks like porphyrin at first glance, but it consists of heavy elements. Quantum chemical calculations show that – contrary to porphyrin – no π electrons are present. Consequently, only weak local ring currents are found, middle, but no ring current all over the entire molecule, in contrast to porphyrin, right (ANGEW. CHEM. INT. ED. 57, 8770-8774 (2018)).
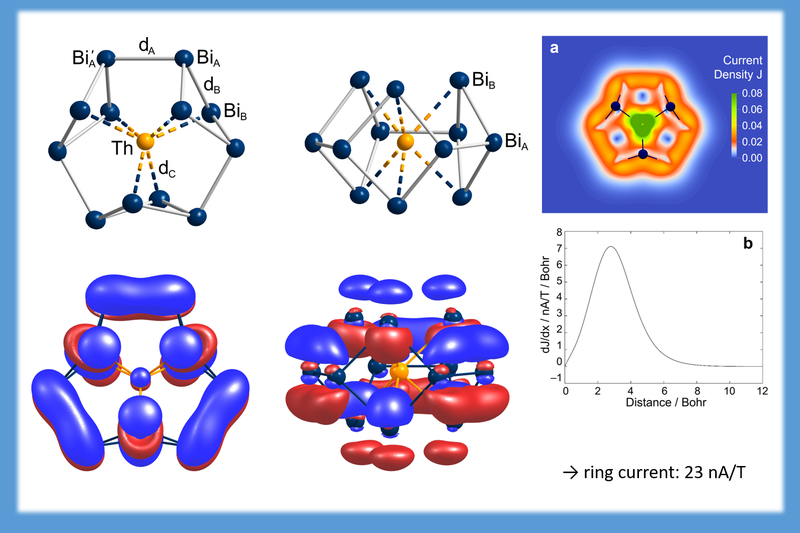
For [Th@Bi12]4- matters are completely different. This system has (only) 2 pi-electrons, but shows a ring current of 23 nA/T. For comparison, in porpyrine 26 pi-electrons are required for a similar ring current strength. The figure shows the cluster in top and side view, the HOMO, which is the only orbital that stays delocalized in all localization procedures, a plot of the absolute value of the current density, and the current profile. (Nature Chemistry 13, 149–155 (2021))
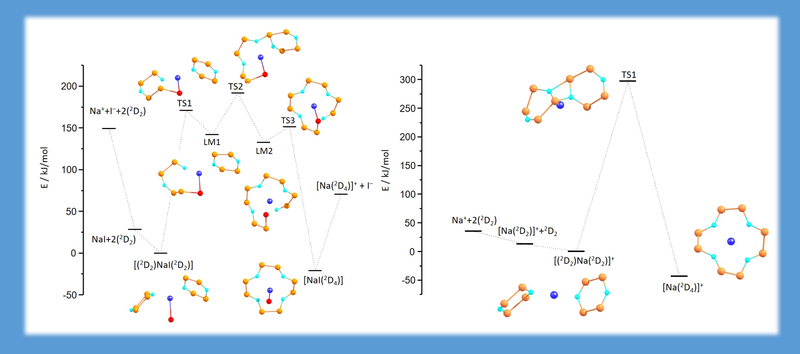
Siloxane units can be effectively activated with the help of a combination of chemically hard cations and iodide or tetraiodidogallate anions. The presence of both is required for acceptable reaction barriers. (ANGEW. CHEM. INT. ED.60,10393(2021)).