Main Content
Method developments in TURBOMOLE
One may characterize the quality of quantum chemical calculations by three parameters. The method used for the treatment of the electron-electron interaction (Hartree-Fock (HF) and post-HF methods or density functional techniques, etc.), the quality of the atomic basis set for expanding the molecular orbitals (double, triple, quadruple zeta, etc.), and, in particular for heavy elements, the quality of the Hamiltonian (non-relativistic, scalar relativistic, two-component, etc).
Our group focuses on the last two aspects and covers the corresponding areas in our program package TURBOMOLE, known for its robustness and efficiency. We implemented
- two-component methods accounting for spin-orbit coupling based on effective core potentials (ECPs) for the ground state (PHYS. CHEM. CHEM. PHYS. 10, 1748-1756 (2008), MOL. PHYS. 111, 2617-2624 (2013)) as well as for excited states (J. CHEM. THEORY COMPUT. 9, 5341-5348 (2013))
- the (one-electron) exact two-component decoupling of the Dirac equation, X2C (J. CHEM. PHYS. 138, 184105 (2013)), J. CHEM. PHYS. 148, 104110 (2018)), the latter in particular for
- the (scalar) relativistic calculation of chemical shielding constants (J. CHEM. THEORY COMPUT. 15, 1028-1043 (2019)).
- NMR coupling constants, nonrelativistic and within the X2C approach (J. CHEM. THEORY COMPUT. 17, 3974-3994 (2021))
- NMR shifts for paramagnetic systems without spin-orbit coupling (J. PHYS.CHEM.A, 125, 9707-9723 (2021)) and with spin-orbit coupling (J. PHYS.CHEM.A, 126, 5050-5069 (2022))
We always very much care for efficiency, in a way that introduced errors are much smaller than changes introduced from altering basis sets or methods. For instance, by employing density fitting and multipole approximation techniques for the Coulomb part as well as sophisticated screening techniques for the Hartree-Fock exchange, chemical shielding constants for glucose chains with several thousand atoms are possible on a single CPU within a day (J. CHEM. THEORY COMPUT. 14, 191-197 (2018)). For the X2C implementations, efficiency is maintained by the employment of the diagonal local approximation (DLU), which is particularly helpful for first and even more for second derivatives of the energy.
These developments are accompanied by the development of error-balanced segmented contracted basis sets for the entire periodic table, for instance,
- for the ECP-based two-component treatments (J. CHEM. PHYS. 133, 174102 (2010)),
- for X2C calculations (J. CHEM. THEORY COMPUT. 13, 3696-3705 (2017)),
- for chemical shielding constants within the X2C method (PHYS. CHEM. CHEM. PHYS. 21, 16658-16664 (2019)).
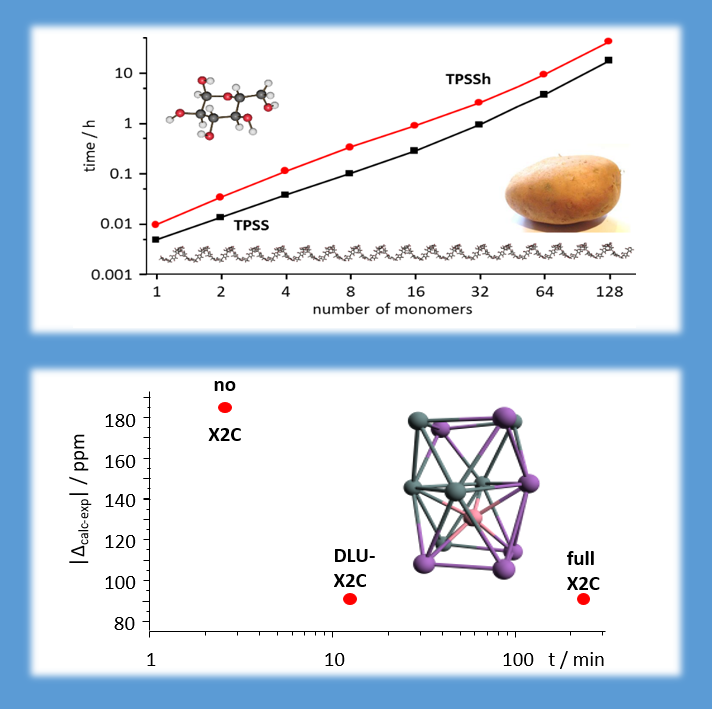
Top: NMR shielding constants of glucose chains calculated with our highly efficient (hybrid-)DFT NMR implementation. Bottom: Accuracy and efficiency of DLU-X2C NMR shifts shown for [Co@Sn6Sb6]3-at level TPSS/x2c-TZVPall/COSMO. Times refer to calculations on a single CPU; all implementation are OpenMP-parallelized allowing for moderate parallel treatments.
Further, we are open for collaborations. For instance, our implementation of the semi-numeric calculation of the Hartree-Fock exchange (J. COMPUT. CHEM. 33, 810-816 (2012)) was used as a very reasonable starting point for the implementation of local hybrid functionals, recently employed for the calculation of chemical shielding constants (J. CHEM. THEORY COMPUT. 16, 931-943 (2020)). Furthermore, the module for the calculation of excitation spectra served as a platform for the pioneering development of GW methods for molecular systems (J. CHEM. THEORY COMPUT. 9, 232-246 (2013)).