Hauptinhalt
2017
ACS APPL. MATER. INTERFACES: "Interfacial Molecular Packing Determines Exciton Dynamics in Molecular Heterostructures: the Case of Pentacene - Perfluoropentacene"
A. Rinn, T. Breuer, J. Wiegand, M. Deck, J. Hühner, R. C. Döring, M. Oestreich, W. Heimbrodt, G. Witte, S. Chatterjee
ACS Applied Materials & Interfaces 9 (48), 42020-42028 (2017), DOI: 10.1021/acsami.7b11118
Dinaphthothienothiophene (DNTT) is a promising new organic semiconductor which combines high charge-carrier mobility with chemical robustness. Although the properties of organic electronic devices are largely determined by the interfaces with electrodes, the interface between DNTT and metals is hardly studied so far. Here, we have examined the interface-structure of DNTT on Ag(111) and multilayers and compared them with films grown on polycrystalline and (111) substrates of silver and gold. In the seed-layer regime, we identify two different interface-structures formed by exclusively flat-lying molecules and a herringbone-arrangement for increased coverages. Combining LEED, STM, and NEXAFS measurements, we have derived precise structure data for both phases and compare their respective adsorption energies derived from thermal-desorption-spectroscopy measurements to explain why both phases are formed. On all examined surfaces DNTT multilayers consist of discrete fibers reflecting a notable dewetting but exhibit different molecular orientations: Molecules are recumbently arranged and form (12-1) oriented islands on the (111) metal substrates, but uprightly oriented on polycrystalline noble metal surfaces. Complementary work-function measurements yield rather similar values for DNTT monolayers on single- and polycrystalline metal substrates, while distinct differences are found for thicker films. Our results appear important for improved transistor geometries, in particular the realization of vertical field-effect-transistors.J. CRYS. GROWTH: "Microstructural Study of Codeposited Pentacene:Perfluoropentacene Grown on KCl by TEM Techniques"

F. Rocio, T. Breuer, G. Witte, K. Volz, K. I. Gies
Journal of Crystal Growth 471, 29-36 (2017), DOI: 10.1016/j.jcrysgro.2017.05.009
The great majority of electronic and optoelectronic devices depend on interfaces between p-type and n-type semiconductors. Finding matching donor-acceptor systems in molecular semiconductors remains a challenging endeavor since structurally compatible molecules may not necessarily be suitable with respect to their optical and electronic properties and the large exciton binding energy in these materials may favor bound electron-hole pairs rather than free carriers or charge-transfer at an interface. Regardless, interfacial charge-transfer exciton states are commonly considered as an intermediate step to achieve exciton dissociation. The formation efficiency and decay dynamics of such states will strongly depend on the molecular makeup of the interface, especially the relative alignment of donor and acceptor molecules. Structurally well-defined pentacene-perfluoropentacene heterostructures of different molecular orientations are virtually ideal model systems to study the interrelation between molecular packing motifs at the interface and its electronic properties. Comparing the emission dynamics of the heterosystems and the corresponding unitary films enables accurate assignment of every observable emission signal in the heterosystems. These heterosystems feature two characteristic interface-specific luminescence channels at around 1.4 and 1.5 eV that are not observed in the unitary samples. Their emission strength strongly depends on the molecular alignment of the respective donor and acceptor molecules, emphasizing the importance of structural control for device construction.APPL. PHYS. LETT.: "Anisotropic thermal expansion in pentacene and perfluoropentacene: Effects of molecular packing motif and fixation at the interface"
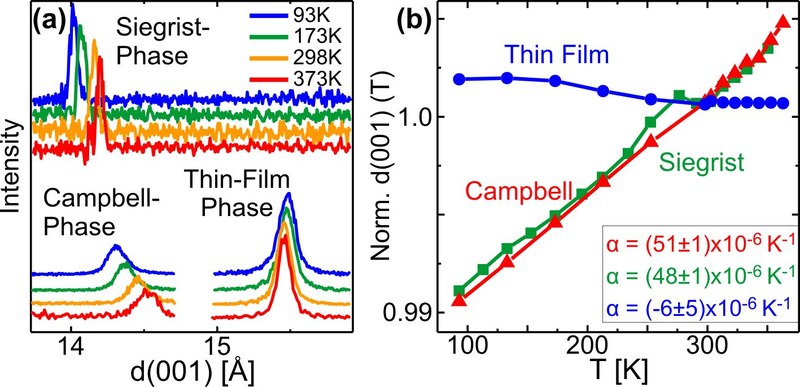
L. von Helden, T. Breuer, G. Witte
Applied Physics Letters 110 (14), 141904 (2017), DOI: 10.1063/1.4979650
Thermal expansion coefficients of molecular solids are typically significantly larger than those of inorganic materials. Since they are furthermore highly anisotropic, the molecular arrangement and consequently the intermolecular orbital overlap strongly depend on temperature, hence also affecting the energetics of optoelectronic excitations and the efficiency of charge transfer processes. Here, we report on the precise determination of the anisotropic thermal expansion coefficients of the organic semiconductor pentacene in its solid state. We compare the thermal expansion coefficients of three different pentacene polymorphs and observe distinct differences between both pentacene bulk polymorphs and the interface-stabilized thin film phase. By comparing epitaxial films with films prepared on weakly interacting, amorphous substrates, we identify a notable influence of the substrate fixation on the thermal expansion in thin pentacene films. Furthermore, the results for pentacene are compared to the thermal expansion of perfluoropentacene, where an exceptionally large vertical thermal expansion coefficient is found in the substrate-mediated π-stacked polymorph. The present study underlines the importance of thermal expansion for the interpretation of temperature-dependent spectroscopic measurements and device characterizations since the notable changes in the unit cell geometries severely affect the intermolecular coupling and thus the excitonic energetics.ACS APPL. MATER. INTERFACES: "Exceptional Dewetting of Organic Semiconductor Films: the Case of Dinaphthothienothiophene (DNTT) at Dielectric Interfaces"
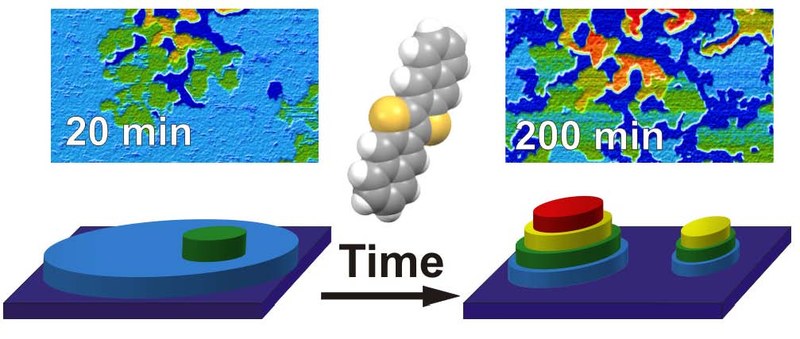
T. Breuer, A. Karthäuser, H. W. Klemm, F. Genuzio, G. Reschel, A. Fuhrich, T. Schmidt, G. Witte
ACS Applied Materials & Interfaces 9 (9), 8384-8392 (2017), DOI: 10.1021/acsami.6b15902
The novel organic semiconductor dinaphthothienothiophene (DNTT) has gained considerable interest because its large charge carrier mobility and distinct chemical robustness enable the fabrication of organic field effect transistors with remarkable long-term stability under ambient conditions. Structural aspects of DNTT films and their control, however, remain so far largely unexplored. Interestingly, the crystalline structure of DNTT is rather similar to that of the prototypical pentacene, for which the molecular orientation in crystalline thin films can be controlled by means of interface-mediated growth. Combining atomic force microscopy, near-edge X-ray absorption fine structure, photoelectron emission microscopy, and X-ray diffraction, we compare substrate-mediated control of molecular orientation, morphology, and wetting behavior of DNTT films on the prototypical substrates SiO2 and graphene as well as technologically relevant dielectric surfaces (SiO2 and metal oxides that were pretreated with self-assembled monolayers (SAMs)). We found an immediate three-dimensional growth on graphene substrates, while an interfacial wetting layer is formed on the other substrates. Rather surprisingly, we observe distinct temporal changes of DNTT thin films on SiO2 and the SAM-treated dielectric substrates, which exhibit a pronounced dewetting and island formation on time scales of minutes to hours, even under ambient conditions, leading to a breakup of the initially closed wetting layer. These findings are unexpected in view of the reported long-time stability of DNTT-based devices. Therefore, their future consideration is expected to enable the further improvement of such applications, especially since these structural modifications are equivalently observed also on the SAM-treated dielectric surfaces, which are commonly used in device processing.PHYS. CHEM. CHEM. PHYS.: Discrete nature of inhomogeneity: The initial stages and local configurations of TiOPc during bilayer growth on Ag(111)
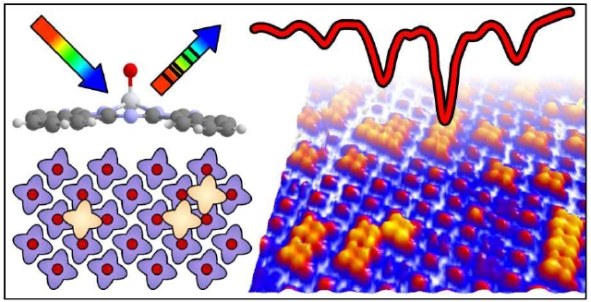
L. Fernandez, S. Thussing, A. Mänz, J. Sundermeyer, G. Witte, P. Jakob
Physical Chemistry Chemical Physics 19 (3), 2495-2502 (2017), DOI: 10.1039/C6CP07922A
The operation of organic optoelectronic devices relies notably on the bulk properties of compound molecular species, but even more so on the influence of interfaces thereof since it is at the interface where elemental electronic processes take place. Their identification and characterization thereby requires that these critical sections of a device are well defined and can be prepared with low defect density. In this context titanyl phthalocyanine (TiOPc) arises as an excellent candidate that reveals the formation of a stable bilayer structure with a characteristic "up-down" molecular arrangement that optimizes the dipole-dipole interaction within the bilayer. In our experimental study, long-range ordered TiOPc bilayers have been grown on Ag(111) surfaces and analyzed using infrared absorption spectroscopy and scanning tunneling microscopy. By monitoring the prominent Ti=O stretching mode in IRAS and identifying local configurations in STM, a microscopic model for the growth of TiOPc bilayers on Ag(111) is suggested. We demonstrate that defect structures within these bilayers lead to characteristic vibrational signatures which react sensitively to the local environment of the molecules. Thermal desorption spectroscopy reveals a high thermal stability of the TiOPc bilayer up to 500 K, which is attributed to hydrogen bonds between oxygen of the titanyl unit and the hydrogen rim of phthalocyanines in the second layer, in addition to contributions arising from the oppositely oriented axial dipole moments and the ubiquitous van der Waals interactions.J. PHYS. CHEM. C.: "Structural and Vibrational Properties of the TiOPc Monolayer on Ag(111)"
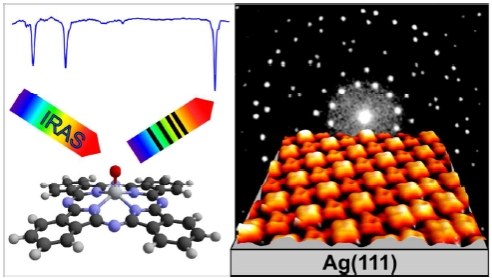
L. Fernandez, S. Thussing, A. Mänz, G. Witte, A. X. Brion-Rios, P. Cabrera-Sanfelix, D. Sanchez-Portal, P. Jakob
Journal of Physical Chemistry C 121 (3), 1608-1617 (2017, DOI: 10.1021/acs.jpcc.6b09701
The evolution of titanyl-phthalocyanine (TiOPc) thin films on Ag(111) has been investigated using IRAS (infrared absorption spectroscopy), SPA-LEED (low energy electron diffraction) and STM (Scanning Tunneling Microscopy). In the (sub)monolayer regime various phases are observed that can be assigned to a 2D gas, a commensurate and a point-on-line phase. In all three phases the non-planar TiOPc molecule is adsorbed on Ag(111) in an oxygen-up configuration with the molecular π-conjugated backbone oriented parallel to the surface. The commensurate phase reveals a high packing density, containing two molecules at inequivalent adsorption sites within the unit cell. Both molecules assume different azimuthal orientations with respect to the Ag(111) substrate which is ascribed to the interplay between the occupation of preferred sites as well as distinct azimuthal orientations and a short ranging Pauli-type repulsion to avoid overlapping of neighboring molecules. At full saturation of the monolayer the latter interaction leads to a further azimuthal reorientation that enables an even higher packing density. Thereby, the plain commensurate order is lost and a point-on-line phase is formed. DFT calculations have been used to study different adsorption geometries of TiOPc on Ag(111). The most stable configurations among those with pointing up oxygen atoms (bridge+, bridgex, topx) seem to correspond to those identified experimentally. The calculated dependence of the electronic structure and molecular dipole on the adsorption site and configuration is found to be rather small