Hauptinhalt
Infrarot-Spektroskopie (FTIR)
Bei Exposition mit Infrarot-Strahlung können bei passender Symmetrie des Auslenkungsmotivs Moleküle und Molekülkristalle zu Schwingungen angeregt werden. Da die Schwingungsenergien von den in der untersuchten Probe vorhandenen Elementen und Bindungen abhängen, ermöglicht die Analyse von Infrarot-Absorptionsspektren eine genaue Untersuchung der chemischen Komposition.
Da die verschiedenen untersuchten Energien nicht dispersiv (mittels eines Monochromators), sondern mit einem Michelson-Interferometer kontinuierlich analysiert werden, verbindet diese Technik typischerweise eine sehr gute Signalempfindlichkeit mit schneller Datenakquisition.
Aufgrund seiner hohen chemischen Sensitivität ist diese Technik überdies sehr gut geeignet, um Kontaminationen und Dekomposition über der Zeit oder unter Zuführung thermischer Energie zu beobachten.
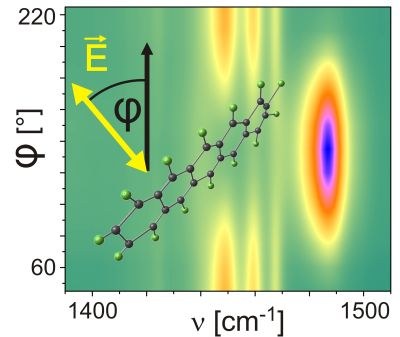
Weitere Möglichkeiten eröffnen sich, wenn die Polarisationsabhängigkeit der Schwingungsmoden ausgenutzt wird: Die verschiedenen Auslenkungsmuster einzelner Vibrationsmoden resultieren in unterschiedlich orientierten assoziierten Dipolmomenten. Zur effizienten Anregung einer Schwingung muss die Polarisation des einfallenden Infrarot-Lichts mit diesem Dipolmoment kompatibel sein, d.h. in die gleiche Richtung zeigen. Dies kann genutzt werden, um über Intensitätsvergleiche von Moden mit verschiedenen Dipolmomenten die Orientierung von Molekülen zu bestimmen.
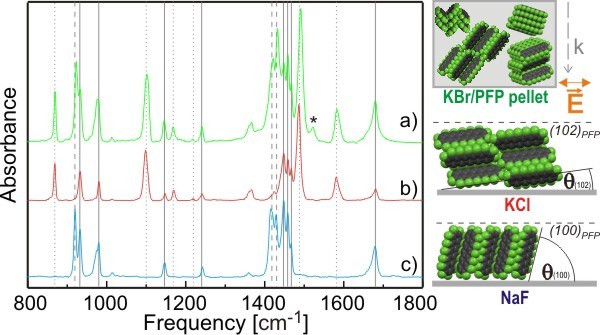
Abbildung 1: Infrarot-Absorption von Perfluoropentacen-Dünnfilmen in unterschiedlicher Orientierung. Die Spektren von liegenden (b) und aufrecht orientierten Molekülen (a) ergeben in Superposition das Spektrum statistisch verteilter Moleküle (c). Je nach Molekülorientierung sind einige dieser Moden sichtbar, andere hingegen nicht. Mehr Details in [T. Breuer et al, J. Phys. Chem. C, 116, 14491 (2012)], siehe insbesondere auch die Supporting Information.
Solche Zuordnungen setzen allerdings die genaue Kenntnis der Auslenkungsmuster voraus. Speziell bei größeren aromatischen Molekülen und Molekülkristallen ist eine solche Zuordnung nicht trivial, da die Schwingungen nicht mehr einer isolierten Bindung zugeordnet werden können, sondern über das gesamte Molekül wirken. Aus diesem Grund müssen für signifikante Analysen die Schwingungsmoden theoretisch oder experimentell zugeordnet werden können. Dies ist durch die Untersuchung wohlgeordneter kristalliner Dünnfilme möglich. Sind diese lateral und vertikal exklusiv orientiert kann durch polarisationsaufgelöste Messungen die Orientierung aller Dipolmomente bestimmt werden.

Abbildung 2: Visualisierung von Schwingungsmoden im PFP-Kristall.
Mehr Details in [T. Breuer et al, J. Phys. Chem. C, 116, 14491 (2012)].
Theoretisch sind solche Beschreibungen durch Anwendung der Dichtefunktionaltheorie (DFT) möglich. Da diese aber systematische Probleme bei der Simulation aufweisen (Schwierigkeiten in der Berechnung der Modenintensitäten, systematische Energiefehler, keine intrinsische Erfassung der stabilisierenden van-der-Waals-Wechselwirkung in Molekülkristallen), benötigen solche Simulationen eine enge Verknüpfung zu entsprechenden Experimenten. Wird dieses geleistet, können aber detailliert Effekte wie der Unterschied zwischen isolierten Einzelmolekülschwingungen und jenen im Festkörper verstanden und sämtliche Auslenkungsmuster anschaulich visualisiert werden.
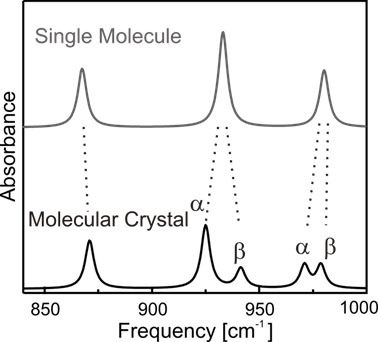
Abbildung 3: Unterschiedliche Absorptionsspektren von Einzelmolekülen und Molekülen im Festkörper. Zu beobachten ist, dass einige der Einzelmolekül-Moden in zwei sog. Davydov-Komponenten aufspalten, andere hingegen nicht. Mehr Details in [T. Breuer et al, J. Phys. Chem. C, 116, 14491 (2012)].
Da unsere Arbeitsgruppe über kein eigenes Infrarot-Spektrometer verfügt, werden IR-Studien in Marburg in Zusammenarbeit mit der AG Oberflächenphysik (→ Oberflächenspektroskopie, Prof. Dr. P. Jakob) durchgeführt.
Beispielhafte Publikationen unserer Gruppe, in denen FTIR genutzt wurde:
- Vibrational Davydov-Splittings and Collective Mode Polarizations in Oriented Organic Semiconductor Crystals.
Tobias Breuer, Mehmet Ali Celik, Peter Jakob, Ralf Tonner, and Gregor Witte
Journal of Physical Chemistry C, 116 (27), 14491-14503 (2012)
Full Text - Selenium as a Key Element for Highly Ordered Aromatic Self-Assembled Monolayers.
Asif Bashir, Daniel Käfer, Jan Müller, Christof Wöll, Andreas Terfort, and Gregor Witte
Angewandte Chemie Int. Ed. 47 (28), 5250-5252 (2008)
Full Text - Vinyl-functionalized gold nanoparticles as artificial monomers for the copolymerization with methyl methacrylate.
Katharina Gries, Mira El Helou, Gregor Witte, Seema Agarwal, Andreas Greiner
Polymer 53 (8), 1632-1639 (2012)
Full Text - A Comprehensive Study of Self-Assembled Monolayers of Anthracenethiol on Gold: Solvent Effects, Structure, and Stability.
D. Käfer, G. Witte*, P. Cyganik, A. Terfort, and Ch.Wöll
J. Am. Chem. Soc. 128 (5), 1723-1732 (2006)
Full Text - Structural Characterization of Organothiolate Adlayers on Gold: The Case of Rigid, Aromatic Backbones.
Claus Fuxen, Waleed Azzam, Ralf Arnold, Gregor Witte, Andreas Terfort, Christof Wöll*
Langmuir 17 (12), 3689-3695 (2001)
Full Text