Hauptinhalt
Hinweise zur Dateninterpretation
Massenbestimmung intakter Proteine:
Die Massenbestimmung für denaturierende Messungen ist sehr genau (messbedingte Abweichung <10 ppm; das bedeutet bei einem Protein mit der Masse 100 kDa sollte die Abweichung max. +/- 1 Da betragen!). Größere Abweichungen bedeuten immer eine Modifikation des gemessenen Proteins bzw. es handelt sich um ein anderes Protein. Besonders häufig findet man Varianten, bei denen das Start-Methionin abgespalten wurde (-131 Da).
Bei nativen Messungen liegen die Primärsignale in einem größeren m/z-Bereich, was dazu führt, dass die messbedingte Abweichung deutlich größer sein kann als bei der denaturierenden Messung. In der Regel geht es hier darum Komplexbildung oder Substrat-/Kofaktorbindung nachzuweisen, so dass es nicht unbedingt auf absolute Genauigkeit ankommt. Letztlich muss hier aber im Einzelfall geschaut werden. Im Zweifelsfall bitte Rücksprache halten.
Proteomics-Applikationen (Proteome Discoverer Daten):
Die Ergebnisse werden in der Regel in From von Excel-Sheets versendet. Zunächst einmal findet sich typischerweise ganz links eine Spalte, die mit einer kleinen "1" überschrieben ist. Darunter befinden sich "+"-Zeichen.
Klickt man auf ein "+"-Zeichen gehen weitere Zeilen zum jeweiligen Eintrag auf, die die identifizierten Peptide und etwaiger zugeordneter Modifikationen enthalten. Gleichzeitig für alle Einträge lassen sich die Unterzeilen für die Peptide öffnen, indem oben links auf die kleine "2" geklickt wird.
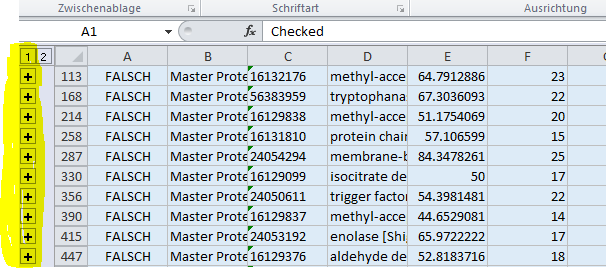
Für eine Übersicht über die Anzahl an Hits sollten diese "Zwischenzeilen" ausgeblendet werden, da ansonsten die Anzahl der Zeilen nicht mit der Anzahl der IDs übereinstimmt. Auf Nachfrage können wir die Daten gerne auch so exportieren, dass entweder nur die reinen Protein-IDs oder alle zugeordneten Peptid-MSMS-Spektren exportiert werden.

Häufig kommt die Rückfrage, was es mit der Spalte "A" und dem angegebenen Wert "FALSCH" auf sich hat. In der Originalsoftware "Proteome Discoverer 2.2" von Thermo Scientific handelt es sich um eine "Check-Box". Zu den selektierten Proteinen lassen sich dann weitere Informationen - wie z.B. die zugeordneten Peptide - anzeigen oder es können selektiv IDs exportiert werden. Da beim Standard-Export nach Excel diese Check-Box typischerweise nicht aktiviert ist wird hier der Wert "FALSCH" ausgegeben. Er ist also schlichtweg für die Dateninterpretation bedeutungslos und auch uns erschließt sich nicht der Sinn, warum er überhaupt softwareseitig mit exportiert wird.
Die "Accession" (Spalte "C") bezieht sich auf die entsprechende Accession-No. aus der verwendeten Proteindatenbank. Wir verwenden typischerweise Uniprot, in Ausnahmefällen auch andere Datenbanken wie z.B. NCBI, oft organismusspezifisch gefiltert, aber manchmal auch eine übergeordnete Taxonomy. U.a. hängt dies auch von den im Auftrag gemachten Angaben, speziell zu den Organismen ab. Wenn einzelne Arbeitsgruppen eigene/andere organismusspezifische Datenbanken mit anderen Accession-No. verwenden, sollte uns der entsprechende FASTA-File wenn möglich noch vor der ersten Auswertung der Daten elektronisch zur Verfügung gestellt werden. Ansonsten ist ein Abgleich der Daten mitunter schwierig. Prinzipiell lassen sich die Daten aber natürlich beliebig oft prozessieren, so dass dies auch noch in einer nachfolgenden Datenbanksuche gemacht werden kann, wenngleich es doppelten Aufwand (vor allem Rechenzeit) bedeutet.
Die "Description" enthält den in dem verwendeten FASTA-File angegebnen Proteinnamen bzw. die -beschreibung.
Die Spalte "Coverage" wird sehr oft überbewertet und falsch interpretiert. Sie ist kein Maß für die Quantität eines Proteins in einer Probe. Es kann sein, dass ein großes Protein, für das warum auch immer nur wenige Peptide identifiziert wurden, eine sehr niedrige Coverage aufweist und ein kleines Protein, mit der gleichen oder auch einer geringeren Anzahl an Peptid-IDs eine sehr hohe, das große Protein aber trotzdem um ein Vielfaches konzentrierter in der Probe vorlag. Selbst unter optimalen Bedingungen kommen 100% Coverage in der Praxis fast nicht vor. Zu beachten ist hierbei auch, dass modifizierte Peptide - posttranslational oder während der Probenvorbereitung - nur dann gefunden werden, wenn diese Modifikationen bei der Datenbanksuche auch berücksichtigt wurden. In der Regel werden bei einer Suche maximal 2-3 weit verbreitete variable Modifikationen berücksichtigt, da ansonsten die Rechenzeiten sehr lang werden und auch die Gefahr falsch positiver zuordnungen steigt.
Sollten bestimmte posttranslationale Modifikationen des Zielproteins bekannt sein oder soll sogar nach bestimmten Modifikationen (z.B. Phosphorylierungen) - auch proteomweit - gesucht werden, so sind diese deshalb unbedingt im Messauftrag anzugeben. Eine nachträgliche erneute Suche unter modifizierten Suchbedingungen ist aber selbstverständlich jederzeit möglich. Liegen alle Peptid-IDs am N- oder C-terminalen Ende kann dies aber auch ein Hinweis auf Prozessierung oder Degradation sein, so dass es sich durchaus lohnen kann die Lage der identifizierten Peptide anzusehen.
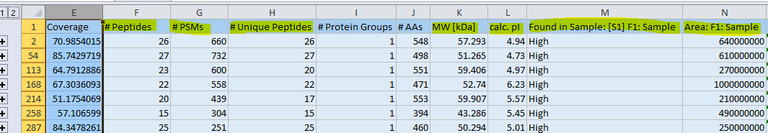
Ein Maß für die Quantität des Proteins in der Probe sind einmal die PSMs (Peptide Spectrum Matches - je höher der Wert, desto mehr MSMS-Spektren wurden diesem Hit zugeordnet) sowie die Abundances (je höher, desto mehr des Proteins war in der Probe bzw. auf der Säule vorhanden). Dabei gilt, dass die PSMs einen schnellen Überblick erlauben, die Abundances aber genauer sein sollten. Einen Blick auf die Quantität zu werfen ist in jedem Fall sinnvoll, da so z.B. Hauptkomponenten eines Pull-Downs von Nebenkomponenten unterschieden werden können. Gerade bei konzentrierten Proben kommt es aufgrund der Sensitivität - auch wenn wir zwischen den Proben einen Leergradienten fahren - teilweise zu unerwünschter Verschleppung. "Verschleppte IDs" lassen sich oft auch anhand der Quantität erkennen. Typischerweise liegen die Intensitäten bei Verschleppung bei ca. 10% (< 1% bei vorherigem Leergradienten) der Ausgangsintensität oder darunter.
Damit ein Hit publizierbar (sprich sicher) ist sollte er min. 3 "Peptides" oder 2 "Unique peptides" beinhalten, auch wenn er in Spalte "M" mit "High" klassifiziert wird. Unterschieden wird hier zwischen "High" "Medium" und "Low". Die standardmäßig eingestellte False Discovery Rate (FDR) liegt bei 1%. Sie wird bestimmt durch den Abgleich der MS-Daten mit einer "Nonsense-Datenbank", die oft durch in silico Revertierung oder aus einem zufälligen Shuffling der Sequencen in einer FASTA-Datenbank erzeugt wird. Das heißt, 1 von 100 Peptid-MSMS-Spektrenzuordnungen ist falsch. Bei zwei "Unique Peptides" liegt die Wahrscheinlichkeit, dass beide falsch sind, schon bei 1:10000. Der Unterschied zwischen Peptides und Unique Peptides ist, dass die Spalte Peptides die Anzahl aller Peptid-IDs beinhaltet, die diesem, aber aufgrund von Sequenzhomologien ggf. auch anderen Proteinen zugeordnet werden können, während sich unter "Unique Peptides" nur solche finden, die genau einmal in dem verwendeten FASTA-File vorkommen. Und dies ist zugleich auch die Einschränkung. Je größer der gesuchte Sequenzraum, desto geringer natürlich die Anzahl der "Unique Peptides". Die Suchmaschinen verwenden ausgeklügelte Algorithmen um trotzdem noch das wahrscheinlichste Protein zu finden. Eine Garantie, dass die Zuordnung mehrfach vorkommender Peptide stimmt, gibt es freilich nicht. Ebenso wie es "False Positives" geben kann, kann es auch "False Negatives" geben. Gerade wenn gezielt ein Protein in einer komplexen Mischung gesucht wird kann es sinnvoll sein eine sehr kleine, spezialisierte Datenbanken zur Suche zu verwenden. Wir verwenden dafür eine "User"-Datenbank, in die wir im Auftrag angegebene FASTA-Files für einzelne Proteine hineinkopieren.
Das wären die wichtigsten Kriterien. Das heißt aber nicht, dass die anderen Hits, die diese Kriterien nicht erfüllen, zwangsläufig falsch sein müssen.
Die Spalte "MW (kDa)" kann man natürlich ebenfalls zu Rate ziehen, insbesondere dann, wenn SDS-Gelbanden analysiert wurden. Hier sollte aber beachtet werden, dass das Laufverhalten auf SDS-Gelen - bedingt durch posttranslationale Prozessierung/Modifikationen oder Degradation der Proteine - aber mitunter deutlich von den Datenbankwerten abweichen kann. Wurde gar ein 2D-Gel mit Trennung in erster Dimension nach den isoelektrischen Punkten durchgeführt, so kann auch der Blick in die Spalte "calc. pI" lohnen, zumindest bei Wackelkandidaten. Aber auch hier ist zu beachten, dass PTMs (u.a. Phosphorylierung) mitunter einen messbaren Einfluss auf den pI haben.
Sollen die Daten zur Label-freien Quantifizierung (relativ oder absolut) herangezogen werden, so wird ebenfalls die Spalte "Area" verwendet. Sowohl für relative als auch für absolute Quantifizierung sind hierfür die Werte in der Spalte "Area" anhand des internen Standards zu normieren. Meist verwenden wir Cytochrom c (Equus caballus) als internen Standard in einer Konzentration von 0.5 µg pro Probe, was dann zur Berechnung der Absolutkonzentrationen verwendet werden kann. Proteome discoverer 2.2 bietet Möglichkeiten mehrere Messungen (z.B. Kontrollen und Proben) quantitativ zu vergleichen. Die gesamte Normierung findet dann schon in Proteome Discoverer statt. Die kann z.B. sinnvoll sein bei Pull-Down-Experimenten, aber auch bei allen anderen Experimenten, bei denen es darum geht Unterschiede zu finden. In solchen Fällen vermerken Sie bitte gleich in den aufträgen, dass ein quantitativer Vergleich über Proben hinweg durchgeführt werden soll. Aber auch nachträglich ist dies natürlich möglich wenn Sie uns kontaktieren und die Wünsche bzw. das experimentelle Design mitteilen. Im Vergleich zu den Standardergebnissen kann dies einiges an Arbeit für die Follow-Up-Datenprozessierung ersparen.
Es kann sein, dass die versendete Tabelle im rechten Teil noch weitere Spalten enthält, z.B. wenn mehr als ein Algorithmus zur Datenbanksuche verwendet wurde (z.B. Sequest und Mascot parallel). Der beschriebene linke Teil der Tabelle enthält dann die zusammengefassten Daten während im rechten Teil die Werte nach Suchmaschinen aufgeschlüsselt sind. Dabei wird es in der Regel eine relativ große Schnittmenge geben, aber auch IDs, die nur mit der einen oder der anderen Suchmaschine erhalten wurden. Das liegt an unterschiedlichen Algorithmen und an der Tatsache, dass es sich um eine automatisierte statistische Auswertung handelt.
Ggf. sind auf der rechten Seite in weiteren Spalten aber auch Informationen zur Zuordnung der jeweiligen ID zu "biochemischen Pathways", "Chromosomenlokalisation", "Ensemble Gene ID", "Gene Symbol", etc. zu finden. Diese Daten werden durch die Zusatzfunktion "Annotation" in Proteome Discoverer 2.2 generiert. Leider ist die Verfügbarkeit dieser Annotationsfunktion abhängig von einer Software Maintenance, die sich Thermo Scientific jährlich finanziell vergüten lässt. Wir verfügen aus Kostengründen derzeit nicht durchgehend über eine Software Maintenance. Wie man zumindest zu einzelnen Einträgen diese Informationen aber auch relativ leicht nachträglich über Uniprot abrufen kann ist hier beschrieben.