Hauptinhalt
Proteomics - Eine Kurzeinführung
Proteomics - eine Kurzeinführung
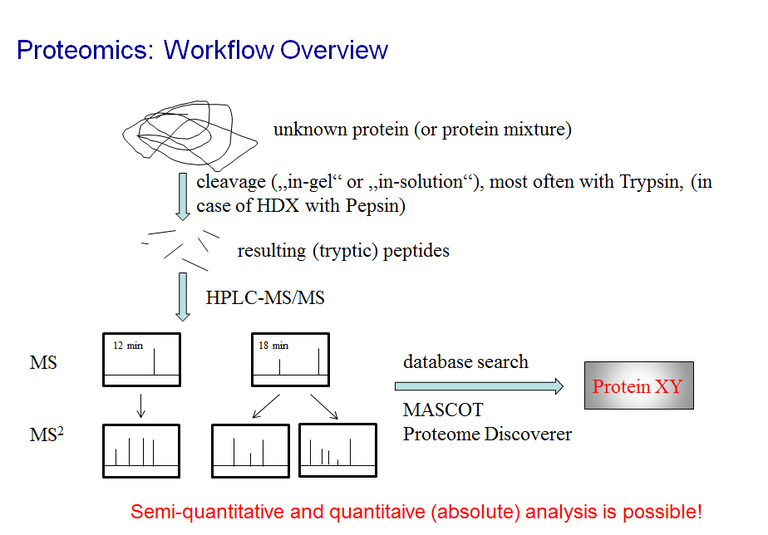
Massenspektrometrische Methoden eignen sich zum Identifizieren von Proteinen und posttranslationalen Modifikationen (PTMs) derselben - entweder aus SDS-Gelbanden bzw. -spots oder auch von Proteinen aus Lösung bzw. mittels FASP. Das Spektrum reicht dabei von der Einzelproteinanalyse (Qualitätskontrolle, Identifizierung von PTMs) bis hin zur vergleichenden bzw. sogar absolut quantitativen Analyse von Komplettproteomen. Dieses Gebiet der massenspektrometrischen Bioanalytik gehört in den Bereich der "omics"-Technologien und wird unter dem Begriff Proteomics zusammengefasst. Voraussetzung ist, dass die Zielproteinsequenzen in Datenbanken (im fasta-Format) vorhanden und für uns zugänglich ist.
Optional - je nach Analysenziel und Erfordernissen - können zunächst sämtliche Cysteine reduziert und alkyliert werden. Die Reduktion erfolgt meist mit DTT (Dithiothreitol) oder TCEP, die nachfolgende Alkylierung oft mit Iodacetamid oder Acrylamid. Die Probe wird vor der massenspektrometrischen Analyse einem proteolytischen Verdau unterzogen. Standardmäßig wird Trypsin, teilweise auch z.B. in Kobination zu LysC verwendet. Trypsin wird gerne verwendet aufgrund seiner hohen Spezifität (schneidet C-terminal von Lysin und Arginin) und einer für die massenspektrometrische Analytik statistisch gesehen optimalen mittleren Peptidlänge. Es sind aber auch andere Proteasen denkbar (Chymotrypsin, AspN, ...). Für schwer lösliche Proben (z.B. Membranproteine) bietet sich der BrCN-Verdau an, der sogar in Gegenwart von hohen Detergenskonzentrationen und/oder in org. Lösungsmitteln durchgeführt werden kann.
Anschließend erfolgt die chromatographische Trennung mittels nanoHPLC und die massenspektrometrische Analyse. Dies funktioniert für "saubere" Gelbanden (Einzelproteine) ebenso wie für komplexe Gemische bis hin zu kompletten Proteomen. Lediglich die Chromatographiebedingungen werden entsprechend modifiziert. Für sehr komplexe Proben kann eine Subfraktionierung oder eine zweidimensionale Chromatographie mit vorgeschalteter Affinitätsreinigung (z.B. Phosphopeptide) oder mittels z.B. Ionentauscher in der ersten Dimension sein.
Die Kombination aus Vorläuferionenmassen (Peptidmassen) und den zugehörigen MSMS-Spektren (beinhalten Sequenzinformation) wird für eine Datenbanksuche verwendet. Derzeit verwenden wir Proteome Discoverer (Version 2.1; Thermo Scientific), Mascot (Version 2.2; Matrix Science - integriert in Proteome Discoverer) sowie die für akademische Zwecke frei zugängliche Software MaxQuant. Je nach Analysenziel wird eine spezialisierte Probenvorbereitung angewendet.
Generell gilt, dass zumindest bei den vergleichenden (quantitativen) Untersuchungen biologische Replikate (mindestens zwei, besser drei oder mehr) verwendet werden sollten. Zusätzlich können technische Replikate gemessen werden, wobei aber der Grundsatz gilt: "Biologische Replikate vor technischen Replikaten". Eine vergleichende Proteom-Studie mit Einzelprobenlaufzeiten (1D) von ca. 4 Stunden kann damit schnell relativ zeitaufwändig - aber auch kostenintensiv - werden. Aus diesem Grund bitten wir darum uns bei Proteomanalysen bereits in der Planungsphase zu konsultieren.